Affymetrix的探针(proble)一般是长为25碱基的寡聚核苷酸;探针总是以perfect match 和mismatch成对出现,其信号值称为PM和MM,成对的perfect match 和mismatch有一个共同的affyID。
CEL文件:信号值和定位信息。
CDF文件:探针对在芯片上的定位信息
affy包是R语言的bioconductor系列包的一个,就一个功能,读取affymetix的基因表达芯片数据-CEL格式数据,处理成表达矩阵!!!
一般我们都是去GEO数据库里面知道找到CEL文件的下载地址~~~比如GSE1438,测了10 young (19-25 years old) and 12 older (70-80 years old) male的样品,然后找差异基因,从GEO数据库我们找到cel文件下载地址是:
ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE1nnn/GSE1428/suppl/GSE1428_RAW.tar
我们是为了讲解affy才下载原始数据的,其实GEO也提供处理好的表达矩阵供下载
下载后压缩到指定目录即可
下载到本地之后就可以用代码读取它了!
library(affy)
dir_cels='D:\\test_analysis\\TNBC\\cel_files'
affy_data = ReadAffy(celfile.path=dir_cels)
eset.mas5 = mas5(affy_data)
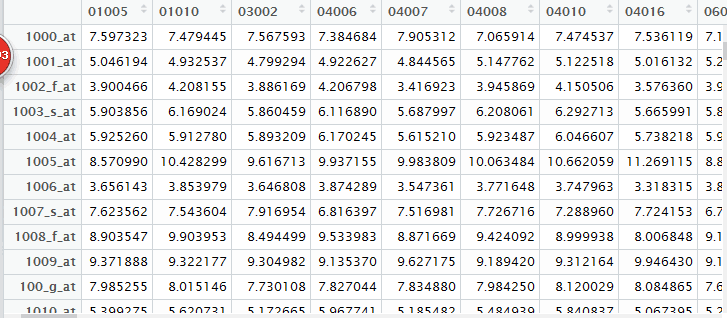
ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE1nnn/GSE1428/matrix/GSE1428_series_matrix.txt.gz
当然这个affy包支持的芯片平台是有限的!
一般是hgu 95系列和133系列~~
其实严格来说,这个芯片得到的表达矩阵,是需要过滤的。
比如像下面的代码:
setwd('../')
library(affy)
dir_cels='GSE34824_RAW'
data <- ReadAffy(celfile.path=dir_cels)
eset <- rma(data)
calls <- mas5calls(data) # get PMA calls
calls <- exprs(calls)
absent <- rowSums(calls == 'A') # how may samples are each gene 'absent' in all samples
absent <- which (absent == ncol(calls)) # which genes are 'absent' in all samples
rmaFiltered <- eset[-absent,] # filters out the genes 'absent' in all samples
54675 features 经过过滤后,剩下 42482 features