仿写fastqc软件的一些功能(下)
文件来自于上面perl代码的输出文件,好像算法有点问题,26G的文件居然处理近一个小时才出数据!
R语言本身自带的画图工具都很丑,懒得说了,可以用ggplot2来重新画一个,不是项目要求没有报酬我就懒得画了,大家面前看看画图原理即可。
a=read.table("meanQ.txt")
看看数据结构如下
> head(a)
V1 V2
1 2 93879
2 3 17800
3 4 25295
4 5 33259
5 6 55685
6 7 84866
plot(a,type='l',col='red',ylab='reads number',xlab='mean quality',main='mean Q distribution')
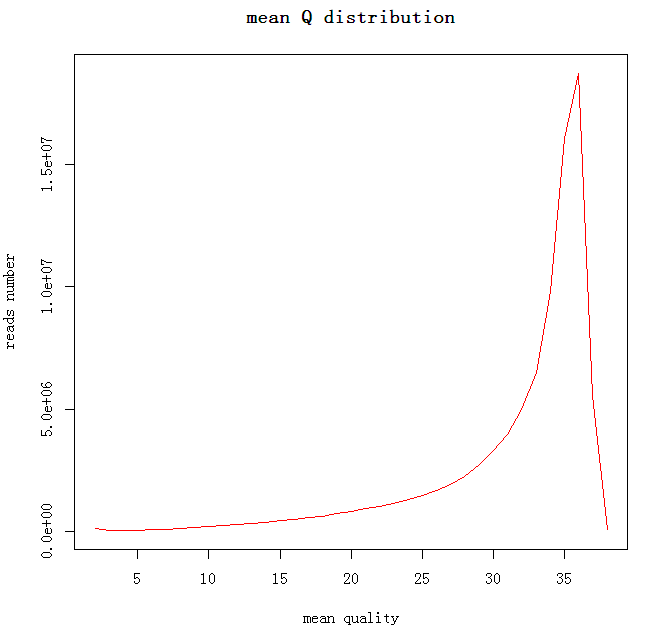
可以看出绝大部分的reads的Q值都在30-35直接,也就是说本次测序挺符合要求的,但是还是需要对那些平均Q20以下的reads过滤掉。
a=read.table('meanGC.txt')
看看数据结构如下
> head(a)
V1 V2
1 0 503
2 1 151
3 2 163
4 3 179
5 4 315
6 5 443
plot(a,type='l',col='red',ylab='reads number',xlab='reads bp',main='GC% distribution')
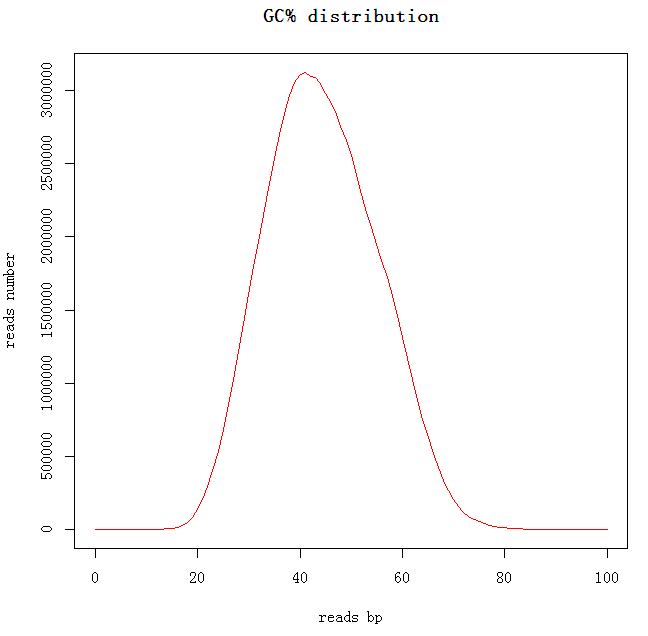
可以看出GC含量的分布看起来挺符合正态分布的,大部分reads的GC含量都是在40%-60%直接
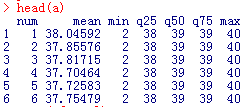
a=read.table('fivenum.txt',header=T)
看看数据结构如下
boxplot(t(a[,3:7]),xlab='reads bp',ylab='Q value',main='mean Q boxplot')
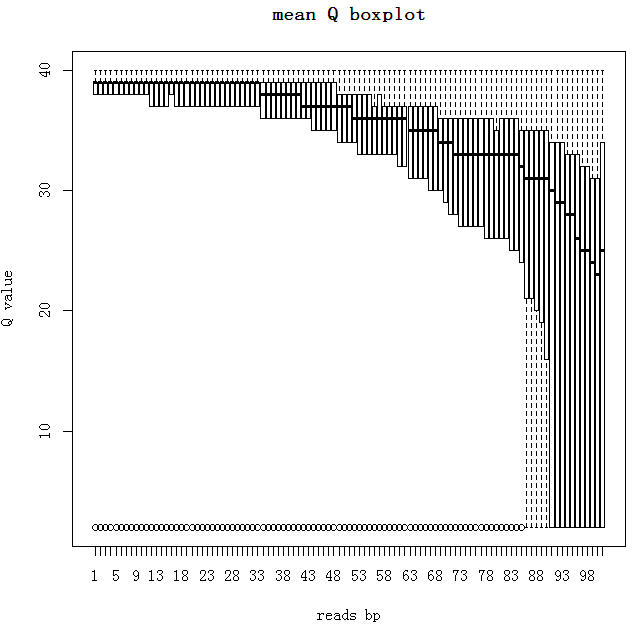
可以看出测序质量从1-100bp过去质量越来越差,但是大部分都是高于Q30,但是88bp之后的碱基测序质量不咋地,可能需要trim掉
对于这个数据还可以画一个图
plot(a[,1:2],type='l',col='red',ylab='Q value',xlab='reads bp',main='mean Q value distribution')
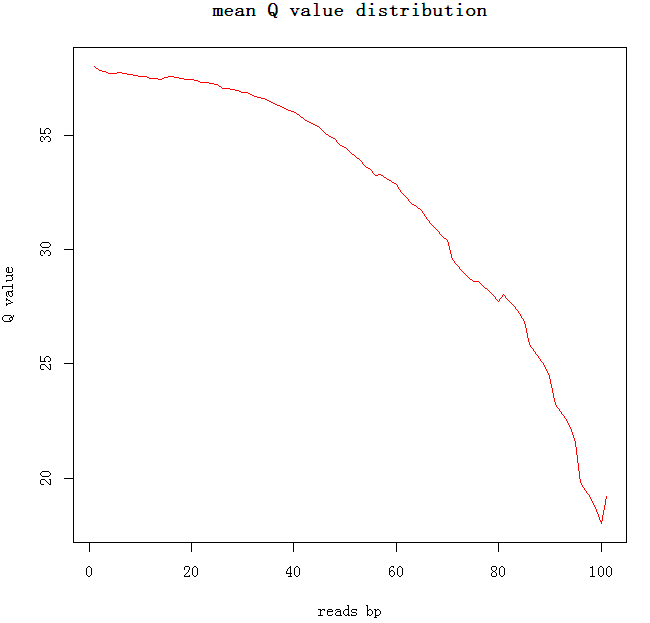
可以看到88bp之后的平均Q值小于30,根据我们的阈值可能要把所有的reads的后面约10个bp的碱基要trim掉