在做这个去除PCR重复reads时候必须要明白为什么要做这个呢?WGS?WES?RNA-SEQ?CHIP-SEQ?都需要吗?随机打断测序才需要?特异性捕获不需要?
搞明白了,我们就开始做,首先拿一个小的单端测序数据比对结果来做测试!
samtools rmdup -s tmp.sorted.bam tmp.rmdup.bam
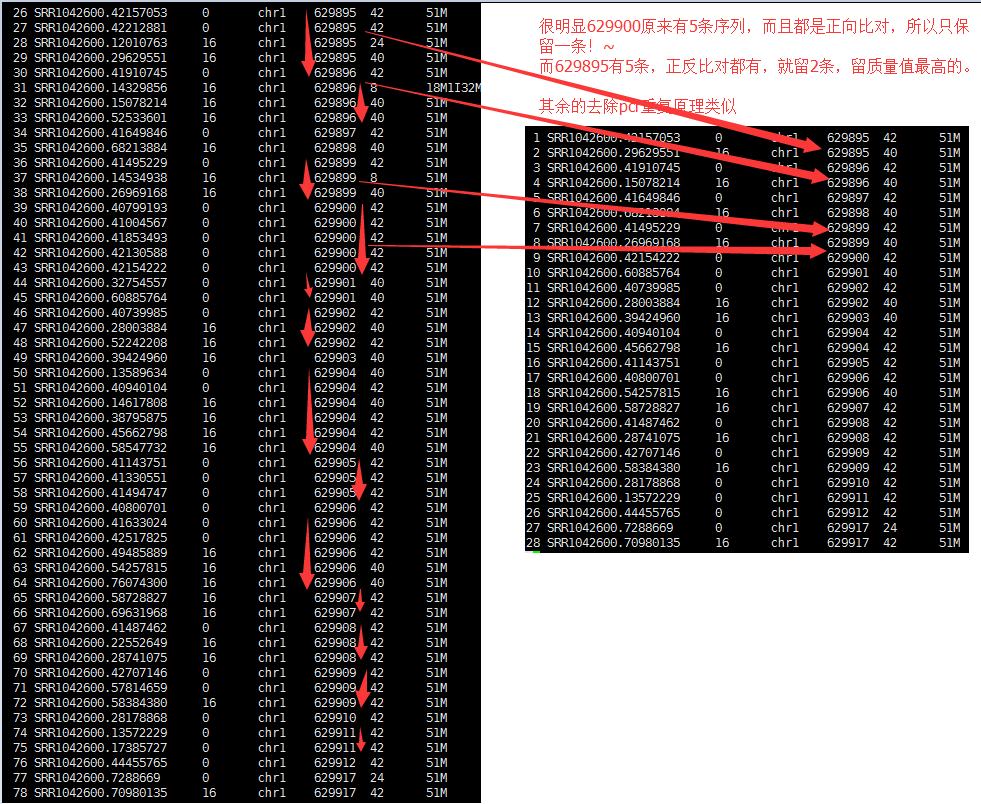
[bam_rmdupse_core] 25 / 53 = 0.4717 in library
我们的测试数据里面有53条records根据软件算出了25条reads都是PCR的duplicate,所以去除了!

samtools rmdup 的官方说明书见: http://www.htslib.org/doc/samtools.html
samtools rmdup [-sS] <input.srt.bam> <out.bam>
只需要开始-s的标签, 就可以对单端测序进行去除PCR重复。其实对单端测序去除PCR重复很简单的~,因为比对flag情况只有0,4,16,只需要它们比对到染色体的起始终止坐标一致即可,flag很容易一致。但是对于双端测序就有点复杂了~
Remove potential PCR duplicates: if multiple read pairs have identical external coordinates, only retain the pair with highest mapping quality. In the paired-end mode, this command ONLY works with FR orientation and requires ISIZE is correctly set. It does not work for unpaired reads (e.g. two ends mapped to different chromosomes or orphan reads).
然后我们再拿一个小的双端测序数据来测试一下:
samtools rmdup tmp.sorted.bam tmp.rmdup.bam
[bam_rmdup_core] processing reference chr10...
[bam_rmdup_core] 2 / 12 = 0.1667 in library
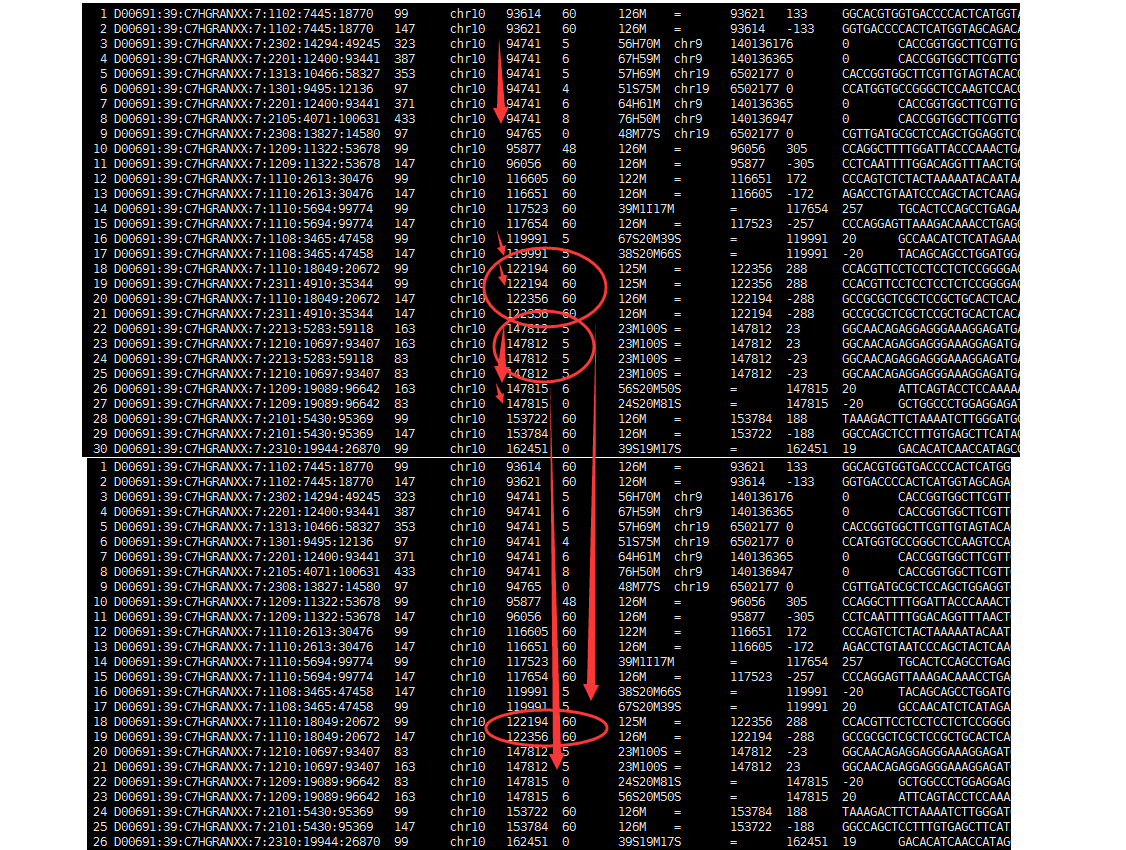
很明显可以看出,去除PCR重复不仅仅需要它们比对到染色体的起始终止坐标一致,尤其是flag,在双端测序里面一大堆的flag情况,所以我们的94741坐标的5个reads,一个都没有去除!

这样的话,双端测序数据,用samtools rmdup效果就很差,所以很多人建议用picard工具的MarkDuplicates 功能~~~
The optimal solution depends on many factors - the consensus seems to be the the picard markduplicates could be the best current solution.
The appropriateness of duplicate removal depends on coverage - one would want to only remove artificial duplicates and keep the natural duplicates.
MarkDuplicates is "more correct" in the strict sense. Rmdup is more efficient simply because it does handle those tough cases. Rmdup works for single-end, too, but it cannot do paired-end and single-end at the same time. It does not work properly for mate-pair reads if read lengths are different.