早在去年九月,我就写个博文说 RNA-seq流程需要进化啦! http://www.bio-info-trainee.com/1022.html ,主要就是进化成hisat2+stringtie+ballgown的流程,但是我一直没有系统性的讲这个流程,因为我觉真心木有用。我只用了里面的hisat来做比对而已!但是群里的小伙伴问得特别多,我还是勉为其难的写一个教程吧,你们之间拷贝我的代码就可以安装这些软件的!然后自己找一个测试数据,我的脚本很容易用的!
其实我最喜欢这样的文章了:http://www.nature.com/nprot/journal/v11/n9/full/nprot.2016.095.html 而且人家还提供了所有的代码,不知道大家怎么还会有疑问的:http://www.nature.com/nprot/journal/v11/n9/extref/nprot.2016.095-S1.zip
人家已经把流程说得清清楚楚了,我还是说一个自己的体悟吧:
软件安装如下:
## Download and install HISAT# https://ccb.jhu.edu/software/hisat2/index.shtmlcd ~/biosoftmkdir HISAT && cd HISAT#### readme: https://ccb.jhu.edu/software/hisat2/manual.shtmlwget ftp://ftp.ccb.jhu.edu/pub/infphilo/hisat2/downloads/hisat2-2.0.4-Linux_x86_64.zipunzip hisat2-2.0.4-Linux_x86_64.zipln -s hisat2-2.0.4 current## ~/biosoft/HISAT/current/hisat2-build## ~/biosoft/HISAT/current/hisat2## Download and install StringTie## https://ccb.jhu.edu/software/stringtie/ ## https://ccb.jhu.edu/software/stringtie/index.shtml?t=manualcd ~/biosoftmkdir StringTie && cd StringTiewget http://ccb.jhu.edu/software/stringtie/dl/stringtie-1.2.3.Linux_x86_64.tar.gztar zxvf stringtie-1.2.3.Linux_x86_64.tar.gzln -s stringtie-1.2.3.Linux_x86_64 current# ~/biosoft/StringTie/current/stringtie
软件使用,我比较喜欢用shell脚本,而且是简单的那种:
while read iddosample=$(echo $id |cut -d" " -f 1 )file1=$(echo $id |cut -d" " -f 2 )file2=$(echo $id |cut -d" " -f 3 )echo $sampleecho $file1echo $file2~/biosoft/HISAT/current/hisat2 -p 4 --dta -x ~/reference/index/hisat/hg19/genome -1 $file1 -2 $file2 -S $sample.hisat2.hg19.sam 2>$sample.hisat2.hg19.log &done <$1
上面这个脚本需要一个3列的输入文件,分别是样本名,read1文件,read2文件,会产生以下的输出文件,sam文件。

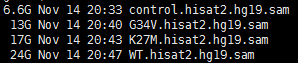
while read iddofile=$(basename $id )sample=${file%%.*}echo $id $samplenohup samtools sort -@ 4 -o ${sample}.sorted.bam $id &done <$1
最新版的samtools已经可以直接把sam文件变成排序好的bam文件啦~~~~

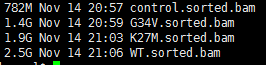
while read iddofile=$(basename $id )sample=${file%%.*}echo $id $samplenohup ~/biosoft/StringTie/current/stringtie -p 4 -G ~/reference/gtf/gencode/gencode.v25lift37.annotation.gtf -o $sample.hg19.stringtie.gtf -l $sample $id &done <$1
stringTie的用法就是这样咯。没什么好讲的
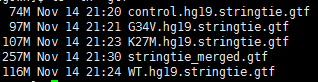

~/biosoft/StringTie/current/stringtie --merge -p 8 -G ~/reference/gtf/gencode/gencode.v25lift37.annotation.gtf -o stringtie_merged.gtf mergelist.txt
while read id
do
file=$(basename $id )
sample=${file%%.*}
echo $id $sample
nohup ~/biosoft/StringTie/current/stringtie -e -B -G $2 -o ballgown/$sample/$sample.hg19.stringtie.gtf $id &
done <$1
我实在讲不下去了,因为真心不用这个东东,我都是拿到了sam/bam文件就直接去counts表达量矩阵了,而count reads数量是非常容易的事情,代码如下
nohup samtools view A.sorted.bam.Nsort.bam | ~/.local/bin/htseq-count -f sam -s no -i gene_name - ~/reference/gtf/gencode/gencode.v25lift37.annotation.gtf 1>A.geneCounts 2>A.HTseq.log &
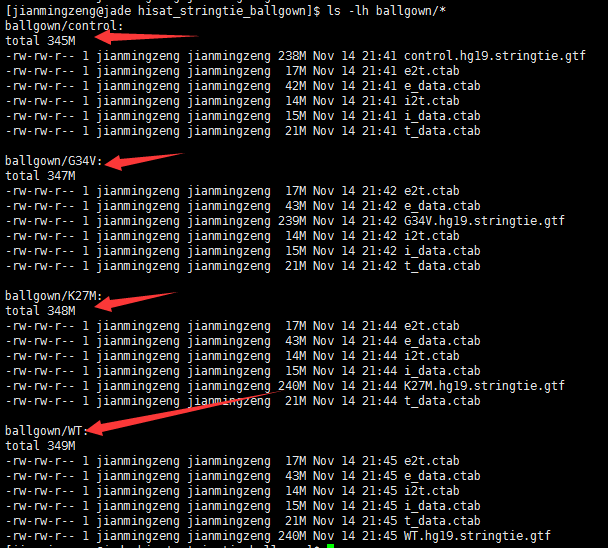
下面的这些文件,导入到R里面用ballgown处理吧,不要在问我这个问题了。