Bioconductor包chimeraviz嵌合RNA可视化
高通量RNA测序已经能够更高效地检测融合转录本,但是融合检测的技术和相关软件通常产生高错误发现率。而一个自动整合RNA数据和已知基因组特征的可视化框架对于结果的检验是有帮助的。2017年发布的一个bioconductor包,chimeraviz就可以做到自动创建嵌合RNA可视化。
支持来自9种不同融合发现工具(deFuse、EricScript、InFusion、JAFFA、FusionCatcher、FusionMap、PRADA、SOAPfuse和STAR-FUSION)的输入。
官网教程
直接在bioconductor可以看到详细说明:https://bioconductor.org/packages/release/bioc/html/chimeraviz.html | HTML | R Script |
下载安装好该R包后,自带一系列的融合基因可视化的测试数据,文件如下:
1.1K Oct 16 22:36 5267readsAligned.bam 96B Oct 16 22:36 5267readsAligned.bam.bai 22K Oct 16 22:36 FusionMap_01_TestDataset_InputFastq.FusionReport.txt 37K Oct 16 22:36 Homo_sapiens.GRCh37.74.sqlite 68K Oct 16 22:36 Homo_sapiens.GRCh37.74_subset.gtf 1.9K Oct 16 22:36 PRADA.acc.fusion.fq.TAF.tsv 32K Oct 16 22:36 UCSC.HG19.Human.CytoBandIdeogram.txt 32K Oct 16 22:36 UCSC.HG38.Human.CytoBandIdeogram.txt 16K Oct 16 22:36 defuse_833ke_results.filtered.tsv 4.6K Oct 16 22:36 ericscript_SRR1657556.results.total.tsv 1.7M Oct 16 22:36 fusion5267and11759reads.bam 57K Oct 16 22:36 fusion5267and11759reads.bam.bai 4.1K Oct 16 22:36 fusioncatcher_833ke_final-list-candidate-fusion-genes.txt 2.1K Oct 16 22:36 infusion_fusions.txt 4.3K Oct 16 22:36 jaffa_results.csv 2.6K Oct 16 22:36 reads.1.fq 2.6K Oct 16 22:36 reads.2.fq 1.0K Oct 16 22:36 reads_supporting_defuse_fusion_5267.1.fq 1.0K Oct 16 22:36 reads_supporting_defuse_fusion_5267.2.fq 3.3K Oct 16 22:36 soapfuse_833ke_final.Fusion.specific.for.genes 2.0K Oct 16 22:36 star-fusion.fusion_candidates.final.abridged.txt
可以看到,所支持的9种融合基因检测工具的示例结果都在这里了,比如我最喜欢的star-fusion的结果节选如下:
#FusionName JunctionReadCount SpanningFragCount SpliceType LeftGene LeftBreakpoint RightGene RightBreakpoint THRA--AC090627.1 27 93 ONLY_REF_SPLICE THRA^ENSG00000126351.8 chr17:38243106:+ AC090627.1^ENSG00000235300.3 chr17:46371709:+ THRA--AC090627.1 5 93 ONLY_REF_SPLICE THRA^ENSG00000126351.8 chr17:38243106:+ AC090627.1^ENSG00000235300.3 chr17:46384693:+ ACACA--STAC2 12 51 ONLY_REF_SPLICE ACACA^ENSG00000132142.15 chr17:35479453:- STAC2^ENSG00000141750.6 chr17:37374426:- RPS6KB1--SNF8 10 43 ONLY_REF_SPLICE RPS6KB1^ENSG00000108443.9 chr17:57970686:+ SNF8^ENSG00000159210.5 chr17:47021337:- TOB1--SYNRG 8 30 ONLY_REF_SPLICE TOB1^ENSG00000141232.4 chr17:48943419:- SYNRG^ENSG00000006114.11 chr17:35880751:- VAPB--IKZF3 4 46 ONLY_REF_SPLICE VAPB^ENSG00000124164.11 chr20:56964573:+ IKZF3^ENSG00000161405.12 chr17:37934020:- ZMYND8--CEP250 2 44 ONLY_REF_SPLICE ZMYND8^ENSG00000101040.15 chr20:45852970:- CEP250^ENSG00000126001.11 chr20:34078463:+ AHCTF1--NAAA 3 38 ONLY_REF_SPLICE AHCTF1^ENSG00000153207.10 chr1:247094880:- NAAA^ENSG00000138744.10 chr4:76846964:- VAPB--IKZF3 1 46 ONLY_REF_SPLICE VAPB^ENSG00000124164.11 chr20:56964573:+ IKZF3^ENSG00000161405.12 chr17:37944627:- VAPB--IKZF3 1 46 ONLY_REF_SPLICE VAPB^ENSG00000124164.11 chr20:56964573:+ IKZF3^ENSG00000161405.12 chr17:37922746:- STX16--RAE1 4 33 ONLY_REF_SPLICE STX16^ENSG00000124222.17 chr20:57227143:+ RAE1^ENSG00000101146.8 chr20:55929088:+
这些结果文件导入R里面统一用import系列函数,比如:
library(chimeraviz)
# Get reference to results file from deFuse
defuse833ke <- system.file(
"extdata",
"defuse_833ke_results.filtered.tsv",
package="chimeraviz")
# Load the results file into a list of fusion objects
fusions <- importDefuse(defuse833ke, "hg19")
## ---- message = FALSE------------------------------------------------------
length(fusions)
基因组全局可视化
soapfuse833ke <- system.file(
"extdata",
"soapfuse_833ke_final.Fusion.specific.for.genes",
package = "chimeraviz")
fusions <- importSoapfuse(soapfuse833ke, "hg38", 10)
# Plot!
plotCircle(fusions)
主要是一个环形图,如下:
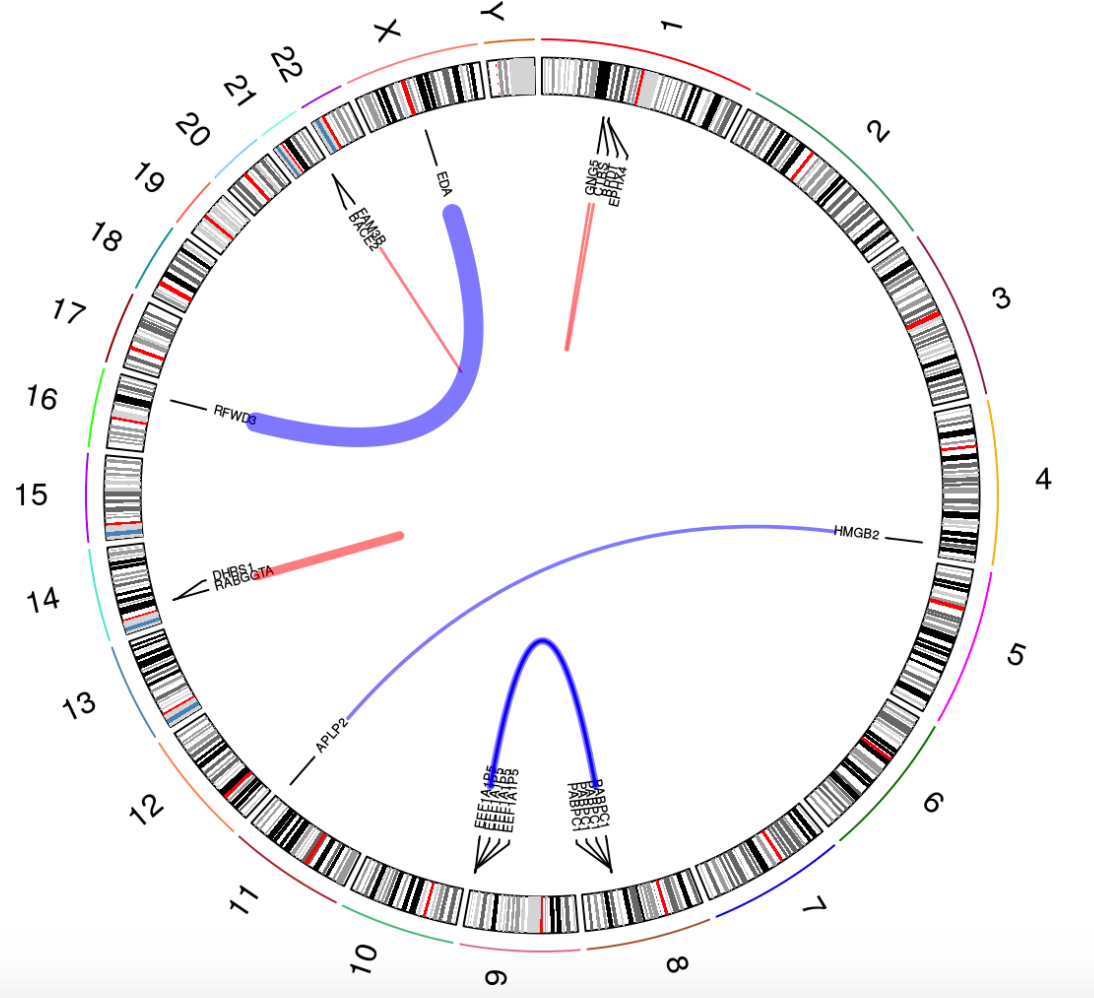
红色条带-染色体内融合,蓝色条带-染色体间融合。
单独可视化某个融合事件
if(!exists("defuse833ke"))
defuse833ke <- system.file(
"extdata",
"defuse_833ke_results.filtered.tsv",
package = "chimeraviz")
fusions <- importDefuse(defuse833ke, "hg19", 1)
# Choose a fusion object
fusion <- getFusionById(fusions, 5267)
# Load edb
if(!exists("edbSqliteFile"))
edbSqliteFile <- system.file(
"extdata",
"Homo_sapiens.GRCh37.74.sqlite",
package="chimeraviz")
edb <- ensembldb::EnsDb(edbSqliteFile)
# bamfile with reads in the regions of this fusion event
if(!exists("fusion5267and11759reads"))
fusion5267and11759reads <- system.file(
"extdata",
"fusion5267and11759reads.bam",
package = "chimeraviz")
# Plot!
plotFusion(
fusion = fusion,
bamfile = fusion5267and11759reads,
edb = edb,
nonUCSC = TRUE)
## ---- echo = FALSE, message = FALSE, fig.height = 5, fig.width = 10, dev='png'----
# Plot!
plotFusion(
fusion = fusion,
bamfile = bamfile5267,
edb = edb,
nonUCSC = TRUE,
reduceTranscripts = TRUE)
这个可视化比较复杂一点,需要融合基因的事件详情,包含两个融合基因的bam片段文件,以及参考基因组的数据库信息。
然后有两种展现方式,一种是基于转录本的融合情况,一种是基于基因
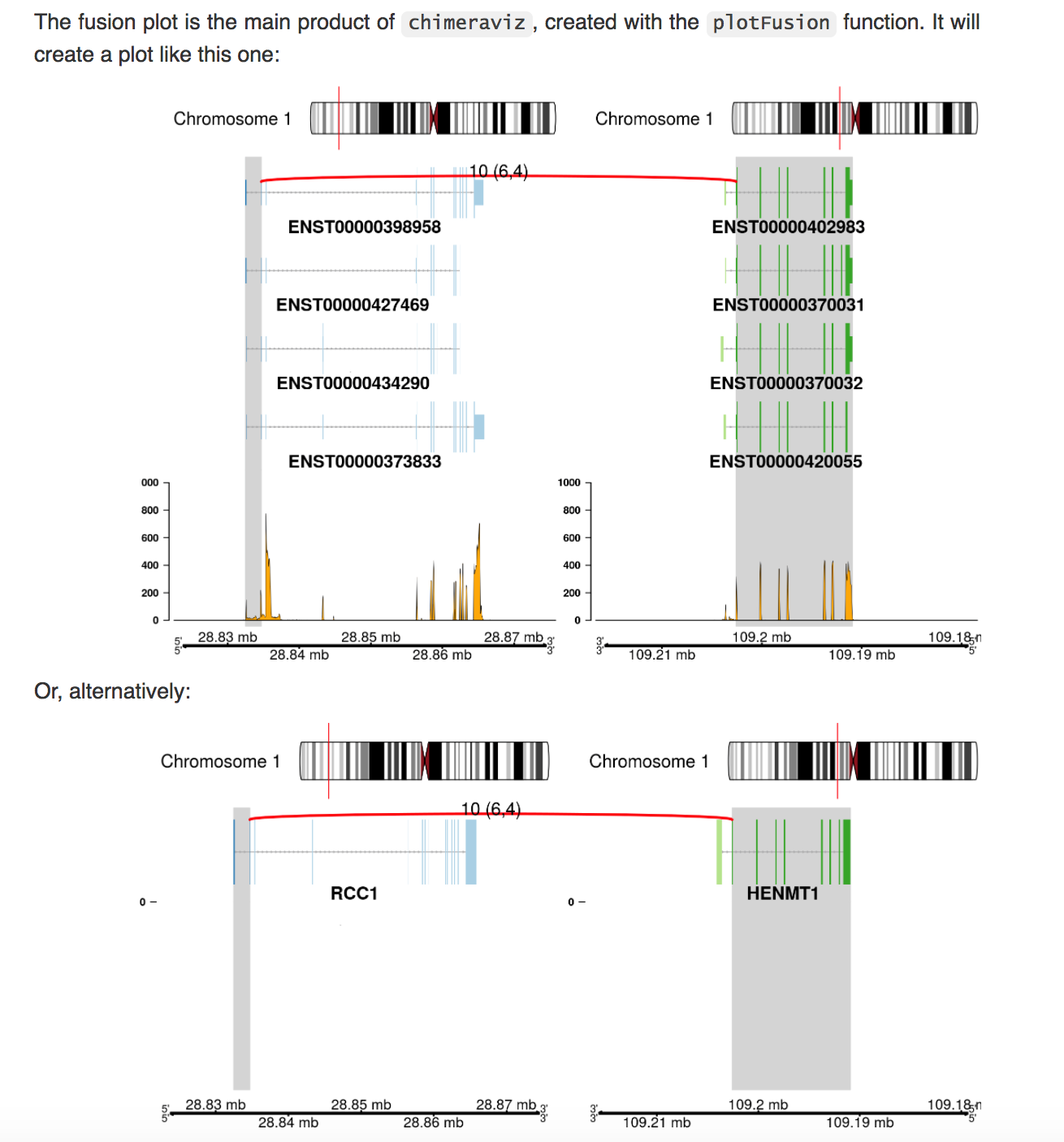
RCC1-HENMT1融合例子。
顶部:显示融合的染色体位置。支持断裂点(红色曲线)的discordant reads数10(其中split的6,spanning的4),注释的转录本及read数图。