赞!
下游分析其实跟产生单细胞转录组测序数据的protocol关系不大,反正都是表达矩阵,但是上游分析的表达矩阵如何获得,这个问题就很大程度上依赖于所采取的单细胞转录组技术了,是否使用spike-in,是否使用UMI, barcode策略是咋样的,等等,都需要考虑。
技术全称是:Illumina Bio-Rad Single-Cell Sequencing Solution
- The ddSEQ Single-Cell Isolator from Bio-Rad encapsulates and partitions single cells into subnanoliter droplets in a disposable cartridge.
- Cell lysis and barcoding occur inside individual droplets for tracking of individual cells throughout the workflow on an Illumina sequencing system.
- This enables transcriptome analysis of hundreds to tens of thousands of single cells in a single experiment.
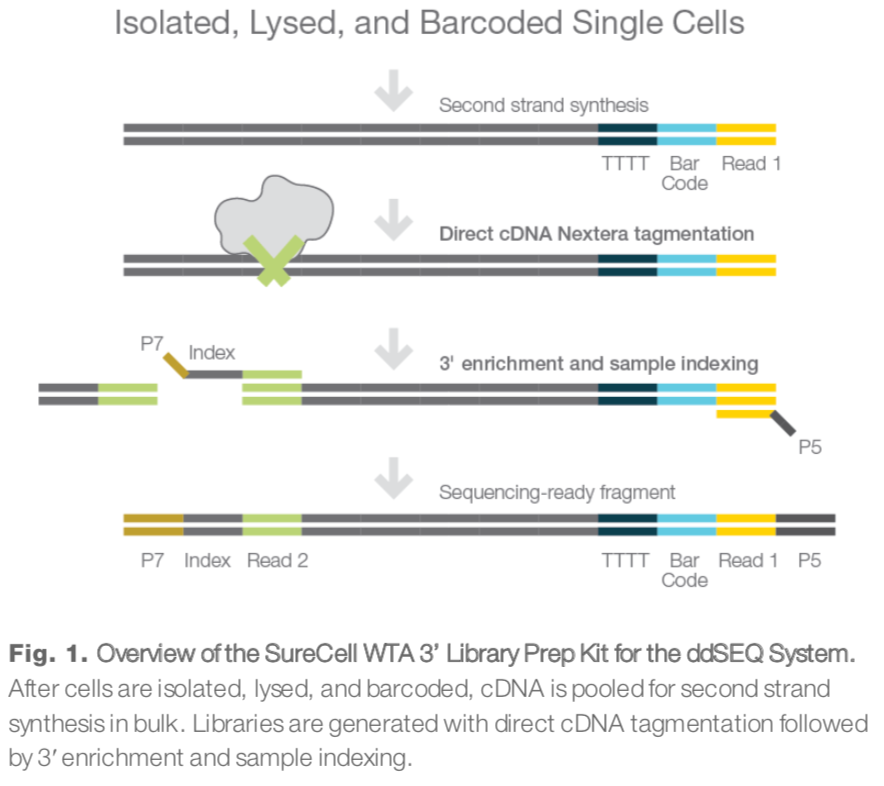
ddseq技术流程
整体上来看,read1包含的都是各种各样的barcodes和UMI,只有read2才是真正的转录本序列,如下所示:
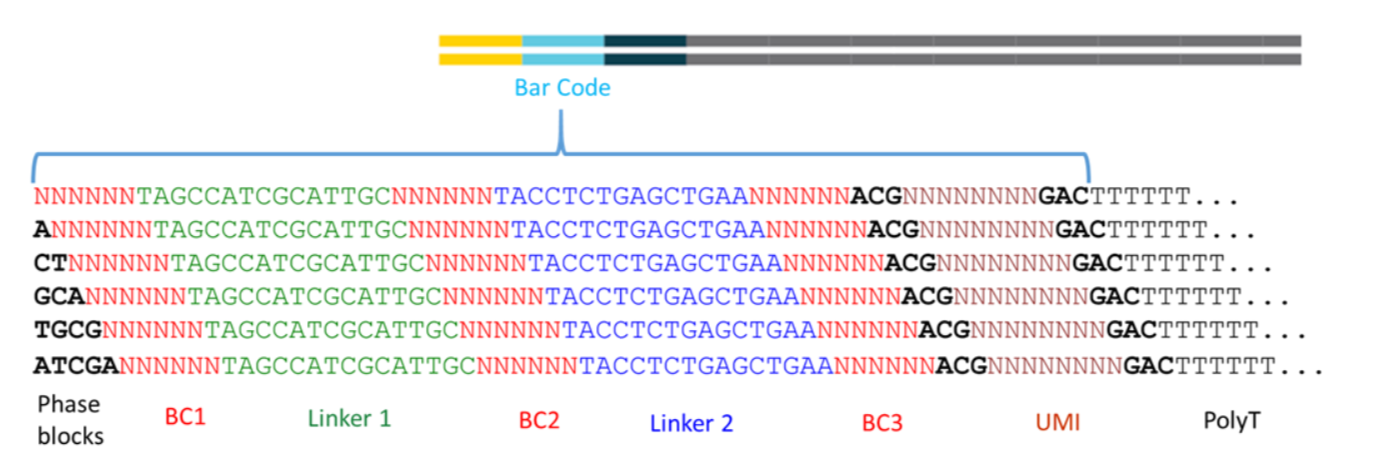
标准的barcodes策略如下:
把序列定位到细胞及转录本
由上图可以看到 read1的序列里面是有两个linker片段是已知的,两个linker把3个6bp的barcode分割开来,第3个barcode和转录本的polyT中间就是 UMI序列。
虽然中间是6bp的barcode,但其实只有 96 possible cell barcode blocks,列表如下:

如果没有开源的已经造好的轮子,那么就需要自己写脚本来进行分析了,主要就是根据barcode来拆分fastq数据到各个细胞。公司的分析流程
其实illumina公司的 BaseSpace SureCell RNA Single-Cell App 可以做数据分析,全套流程都有,如下:
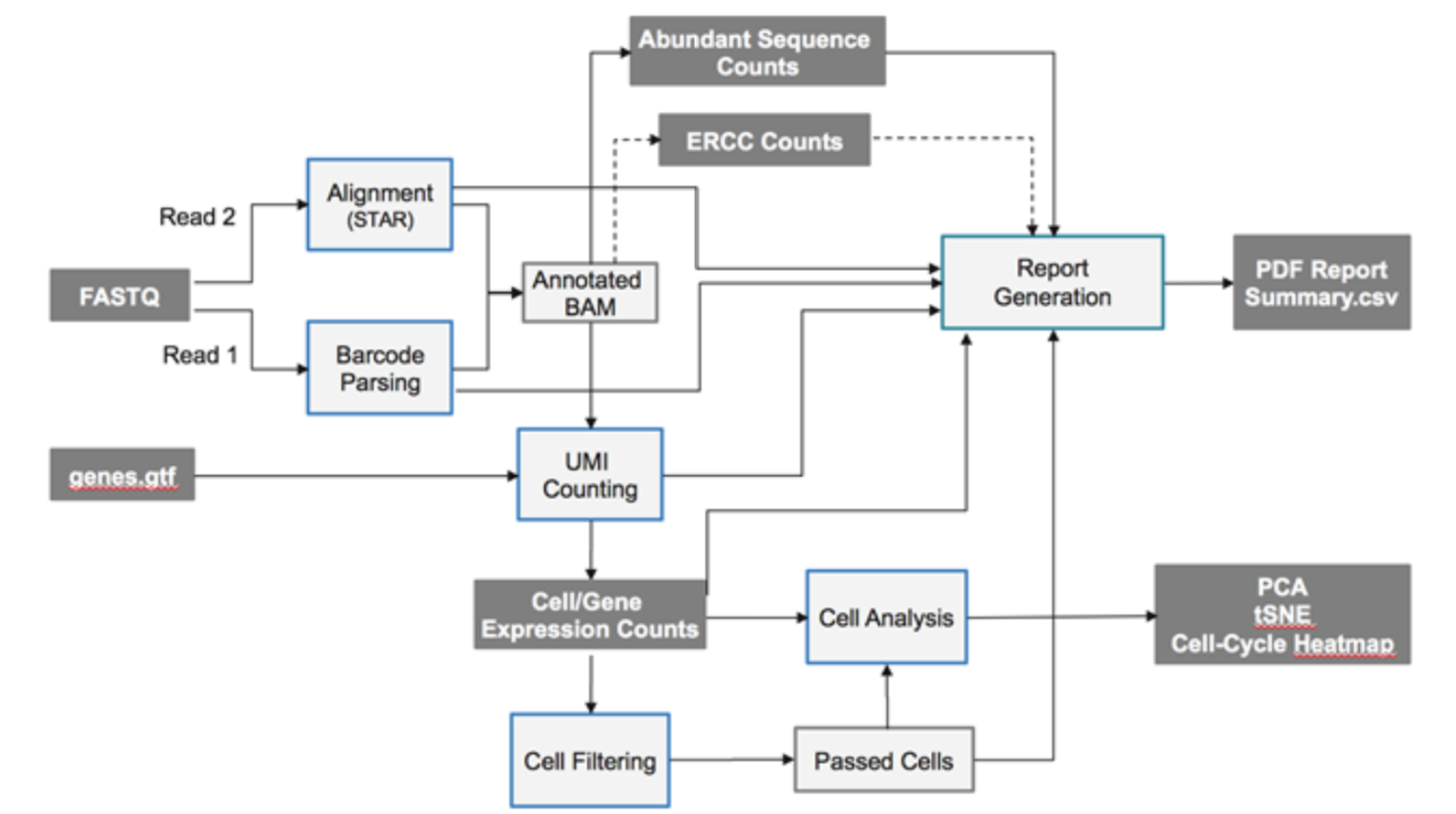
因为只有reads2是转录本的序列,所以用STAR比对到参考基因组的时候把测序数据当做是单端测序即可。
理论上可以得到如下的表达矩阵:

项目数据的评价指标
主要是看下面这些指标
-
The # valid barcodes is provided in the first table of the analysis results. This metric shows how well read 1 performed, and is essential for identifying cells.
-
The # aligned reads shows how well read 2 aligned to genes in the selected reference genome.
-
The percent of reads aligned to unique genes shown in the report provides insight into the read utilization for the sample. This metric represents the number of reads with valid barcode that passed QC and aligned to unique genes with a mapping quality of > 11.
- Cells passing filter represents the number of cells with a UMI count above the knee threshold. This will exclude cells with few UMIs (low RNA content or low library efficiency).
- Median genes detected in cells passing filter + Median UMIs per cell passing filter.
自己写脚本拆分测序文件到各个细胞
待续: