文章发表于:Cell. 2018 May
背景
TNBC是最恶性的乳腺癌,目前通行治疗策略是新辅助化疗,但是会有30%–50%的TNBC病人抵抗,因为肿瘤异质性,很大的一个疑问是化疗耐受后的肿瘤里面的突变是之前就存在呢还是治疗途中产生的。
TNBC的异质性一直是热点,主要有3种研究策略:
- Deep-sequencing studies (Balko et al., 2012, Balko et al., 2014, Shah et al., 2012)
- multi-region sequencinganalysis (Yates et al., 2015)
- single-cell sequencing studies (Gao et al., 2016, Navin et al., 2011, Wang et al., 2014)
化疗耐受后的肿瘤里面的突变是之前就存在呢还是治疗途中产生的这个研究在各种癌症里面有数据,结论不一致。 - acquired resistance (Ding et al., 2012, Kim et al., 2015, Kolodziejczyk et al., 2015, Patch et al., 2015)
- adaptive resistance (Ding et al., 2012, Kurtova et al., 2015)
数据有: - In acute myeloid leukemia, whole-genome sequencing identified different modes of clonal evolution, with some patients acquiring relapse-specific mutations and others selecting minor clones (Ding et al., 2012).
- In high-grade serous ovarian cancer, platinum-based chemotherapy induced new somatic mutations, consistent with acquired resistance (Patch et al., 2015), while resistance to cytotoxic chemotherapy in bladder cancer was associated with the selection of pre-existing subpopulations (Kurtova et al., 2015).
- In glioblastoma, treatment with temozolomide induced many new mutations in post-treatment tumor samples, consistent with an acquired model of therapy resistance (Kim et al., 2015, Kolodziejczyk et al., 2015).
实验设计
作者们一开始的实验设计就很清晰,知道自己要做什么,就是为了探索为什么同样的TNBC病人会对新辅助化疗产生不同的反应呢?化疗耐受后的肿瘤里面的突变是之前就存在呢还是治疗途中产生的?
整体实验设计如下,每个步骤的数据分析也都很清晰,更重要的是作者的数据分析结果完美的解释和回答了他们之前的假设。
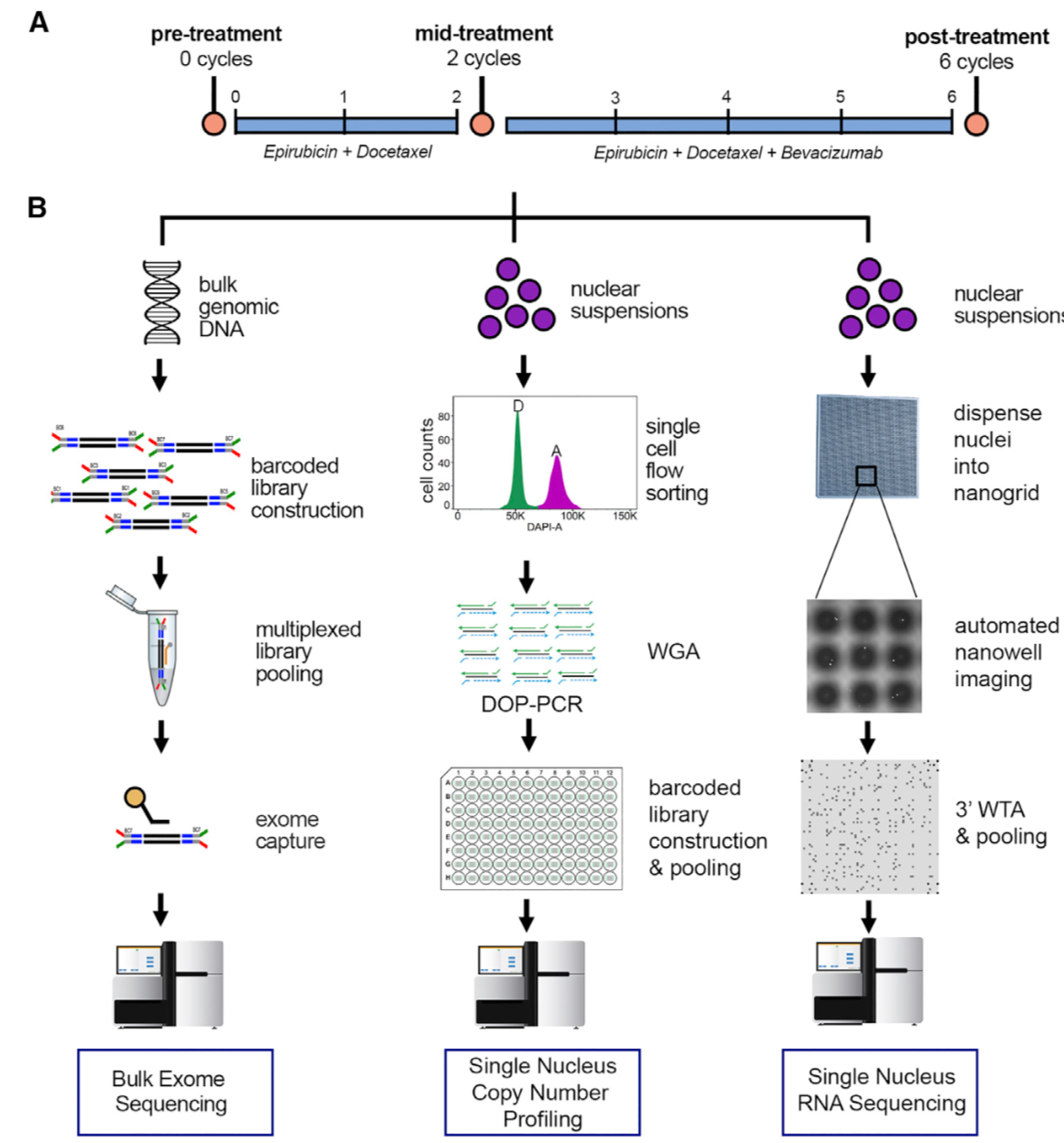
肿瘤外显子测序
包括20 TNBC病人的bulk WES测序数据,blood的对照以及新辅助化疗前后肿瘤,还有部分治疗中的肿瘤测序数据。
11 WXS 2cycleschemo 21 WXS blood 19 WXS operative 21 WXS pre走的是肿瘤外显子的标准分析流程,20 TNBC病人恰好是10个新辅助化疗受益,10个抵抗。
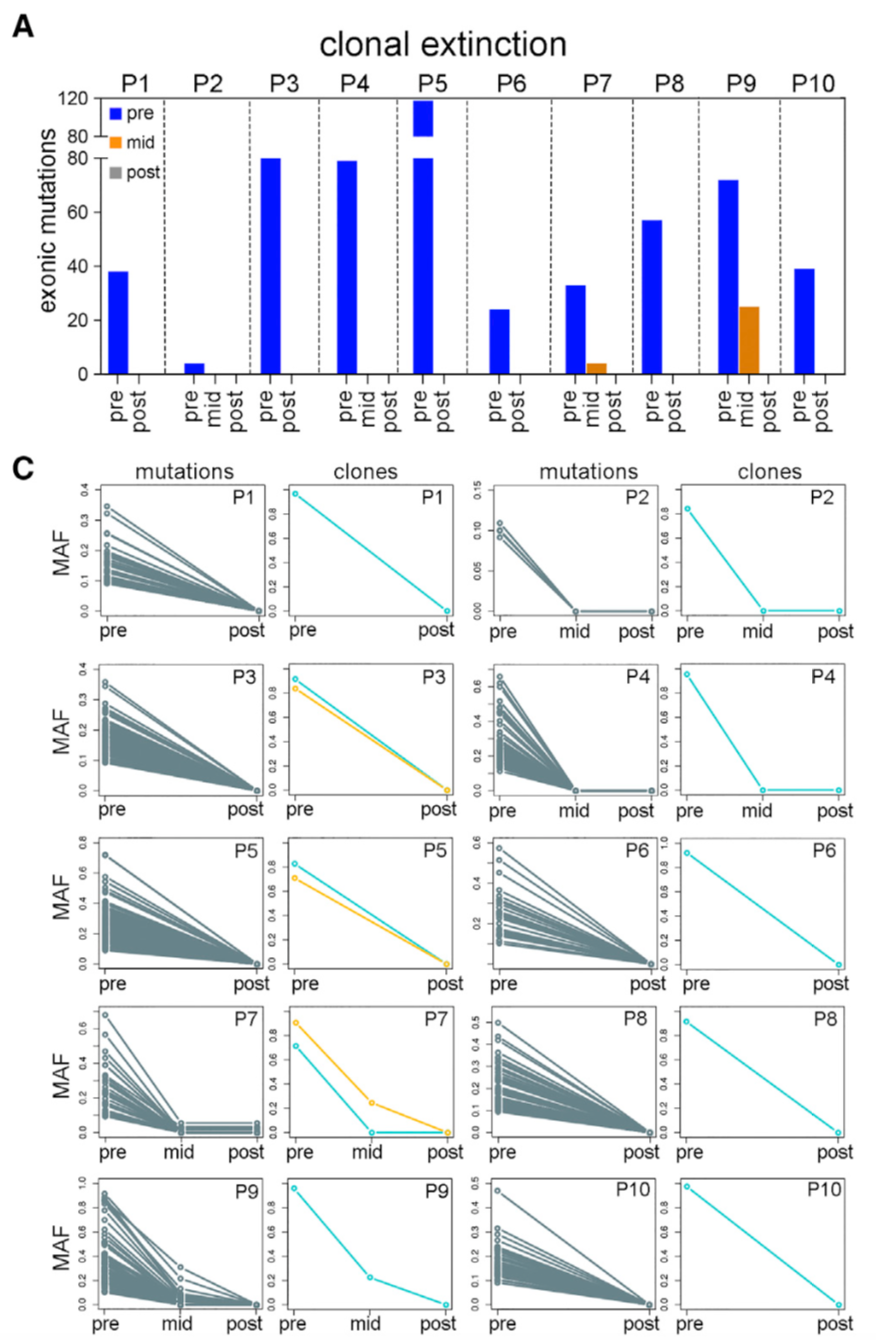
下面是10个新辅助化疗受益,肿瘤消失的病人
可以看到下面的10个病人的化疗之前的WES数据里面的很多somatic mutation都在化疗后取样的WES数据里面找不到了:
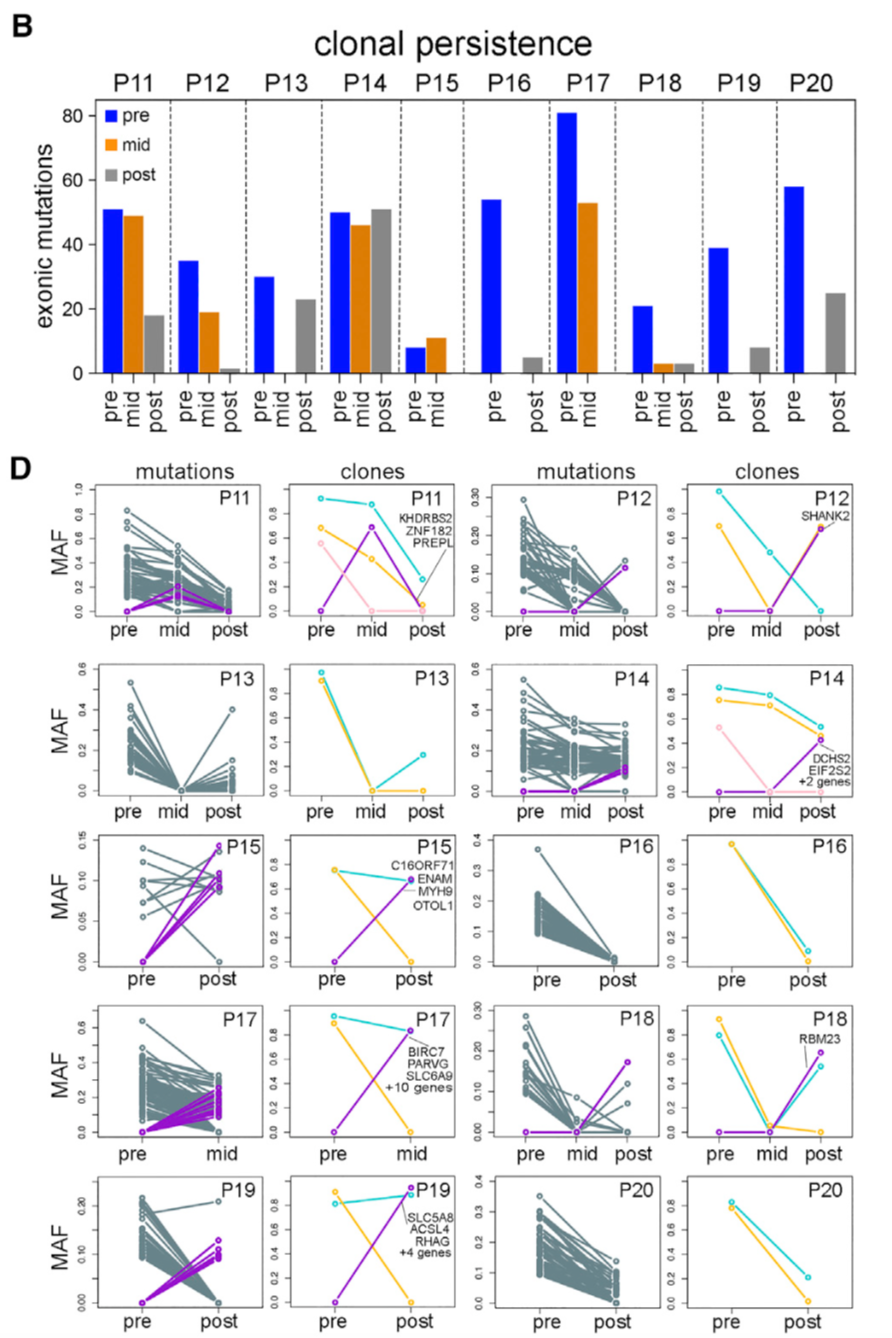
可以看到下面的10个病人的化疗之前的WES数据里面也是有很多somatic mutation都在化疗后取样的WES数据里面找不到了,但同时有很多突变在化疗前没有,但是在化疗后却出现了!!!
高深度测序解决突变是否存在于化疗前
因为作者采用的bulk的肿瘤外显子测序平均测序深度是188X,认为对somatic mutation来说,置信度仍然不高,所以采取了高深度测序,来探索那些新辅助化疗耐受的病人化疗后新出现的 somatic mutation是因为在化疗前的肿瘤WES里面由于测序深度(肿瘤克隆比例极低)不够导致没有被发现呢,还是本身就之前没有,而是化疗诱导产生的。
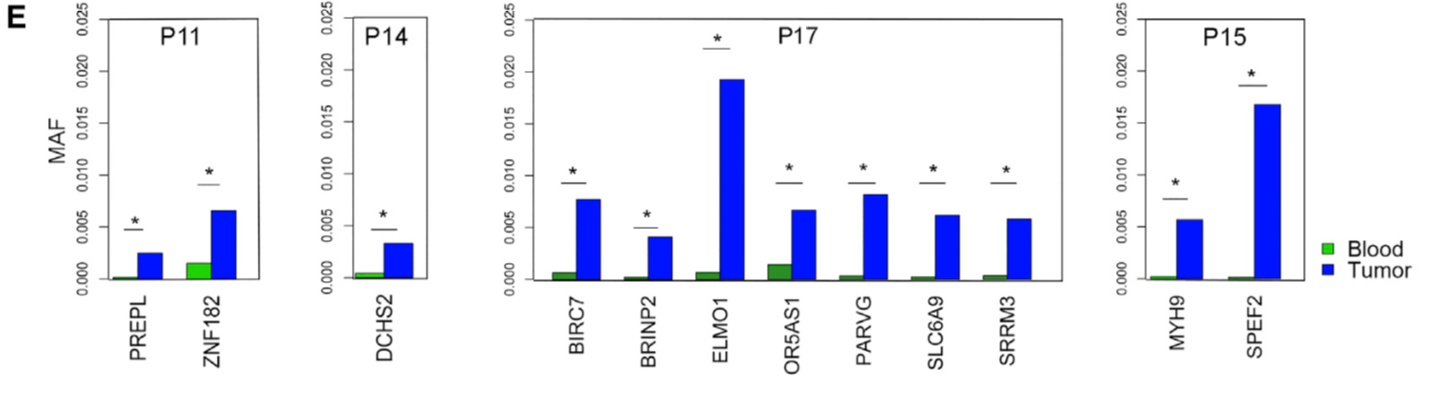
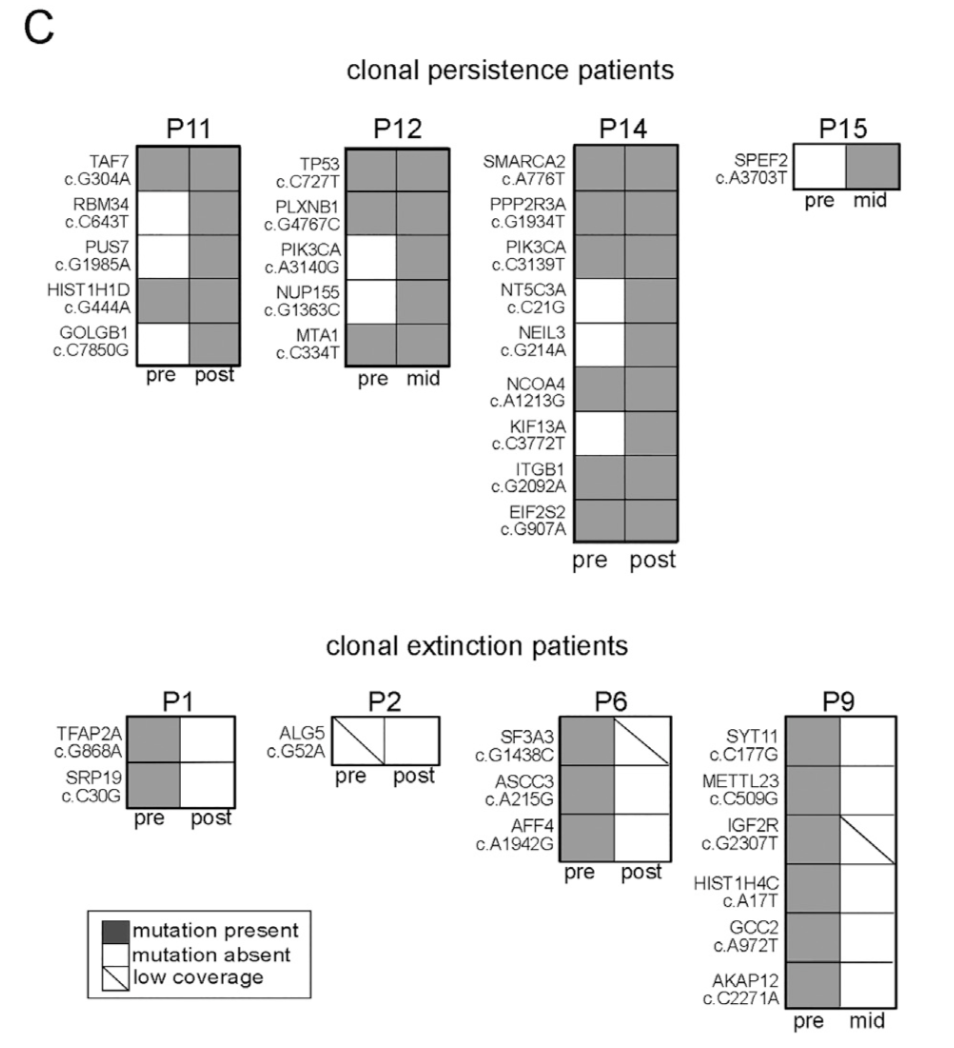
值得注意的突变,作者也单独列出来了:
单细胞DNA测序
测序是:single-nucleus sequencing (SNS) (Gao et al., 2016, Navin et al., 2011) on 900 single cells from matched longitudinal samples of 8 TNBC patients,包括:
- four clonal extinction patients (P1, P2, P6, P9)
- four clonal persistence patients (P11, P12, P14, P15)
使用FACS挑选细胞,全基因组测序,深度0.1X即可,只探索CNV信息。
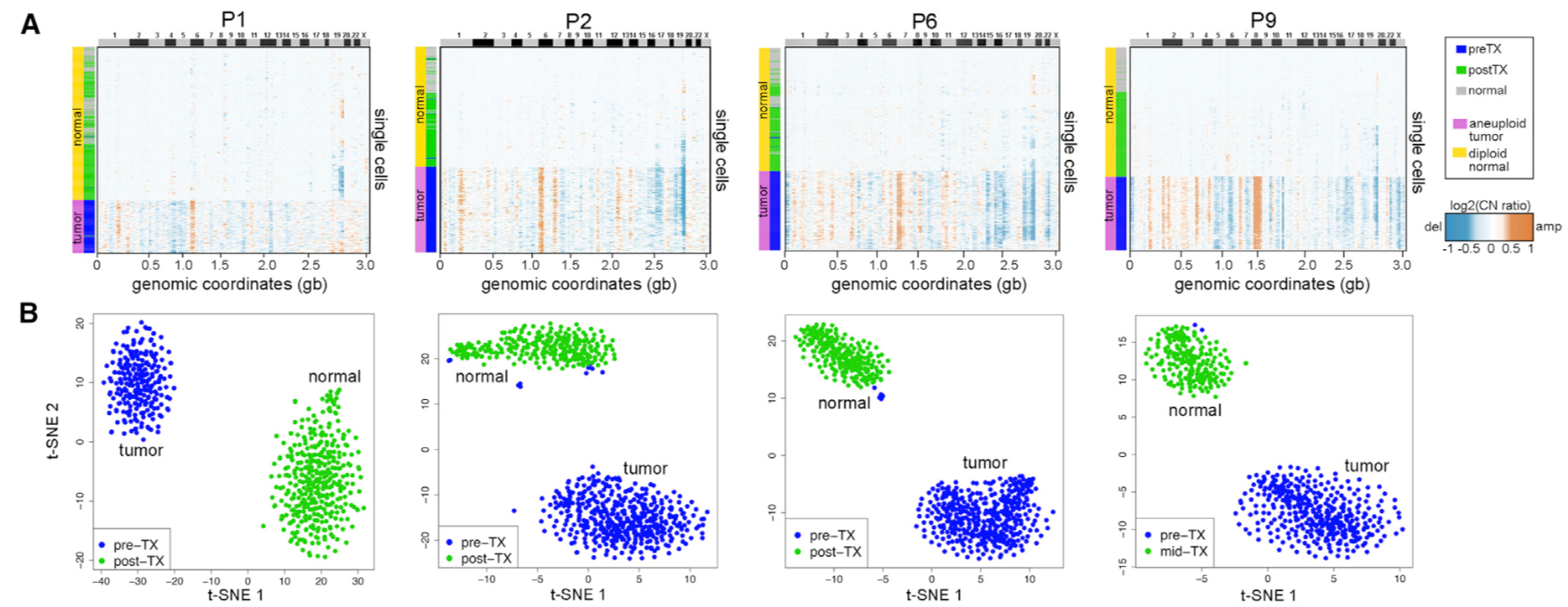
对于那些新辅助化疗受益的病人来说,很明显化疗前后的单细胞DNA测序数据的CNV可以把它们清晰的分开,化疗前的细胞有着癌细胞的特性,但是化疗后的基本上都是正常细胞。
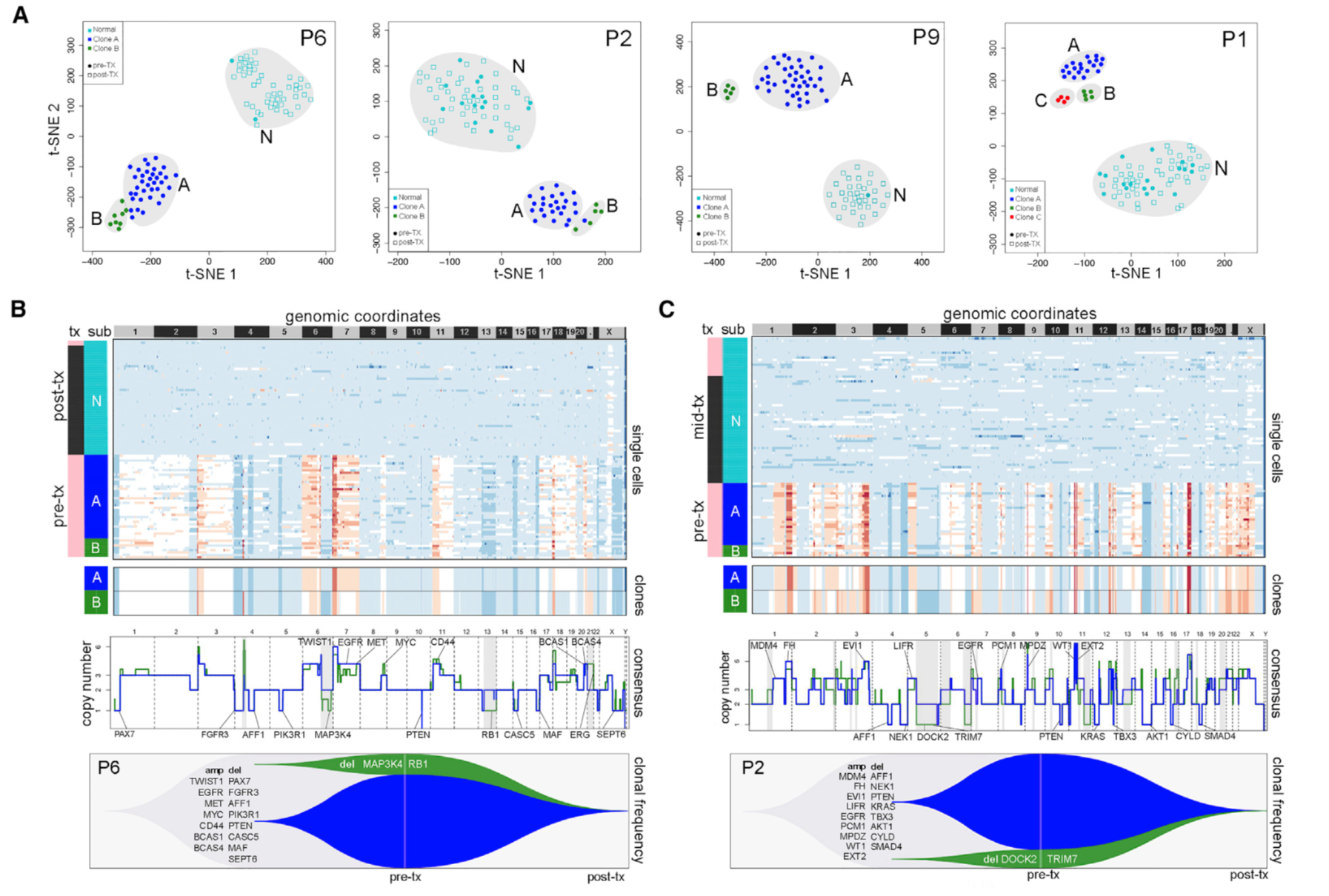
对于那些新辅助化疗抵抗的病人来说,同一个病人的化疗前后的单细胞都表现为癌细胞的特性,而且有着不同的克隆,说明了这些病人在化疗的过程中癌症有着一定程度的进化,产生了新的突变。

作者在2016年就做过 sequenced 1,000 single cells from tumors in 12 patients with TNBC ,数据是: SRP064210 for tumors T1–T10 and SRA018951 for tumors T11 and T12 , 也是根据CNV信息看克隆结构。单细胞转录组测序
测序方法是:a high-throughput nanogrid single-nucleus RNA-sequencing (SNRS) method (Gao et al., 2017). 纳入的病人是:
- 3,370 single nuclei isolated from two matched longitudinal samples per patient from the four clonal extinction patients (P1, P2, P6, P9).
- performed SNRS on ∼400 nuclei from each matched time-point per clonal persistence patients (P11, P12, P14, P15)
还利用了作者 Nat. Commun, 8 (2017) , 文章里面的240 diploid normal breast cells作为单细胞转录组CNV分析的对照,数据也是在GEO, SRP095350
每个病人的细胞个数如下:91 KTN1020 184 KTN102OP 254 KTN1260 481 KTN126OP 478 KTN1290 407 KTN129OP 217 KTN1320 450 KTN1322 478 KTN1520 415 KTN152OP 406 KTN3020 474 KTN3022 285 KTN6150 227 KTN6152对于那些新辅助化疗受益的病人来说,从单细胞转录组的CNV信息很清楚的可以看到他们治疗后癌细胞含量极低了。
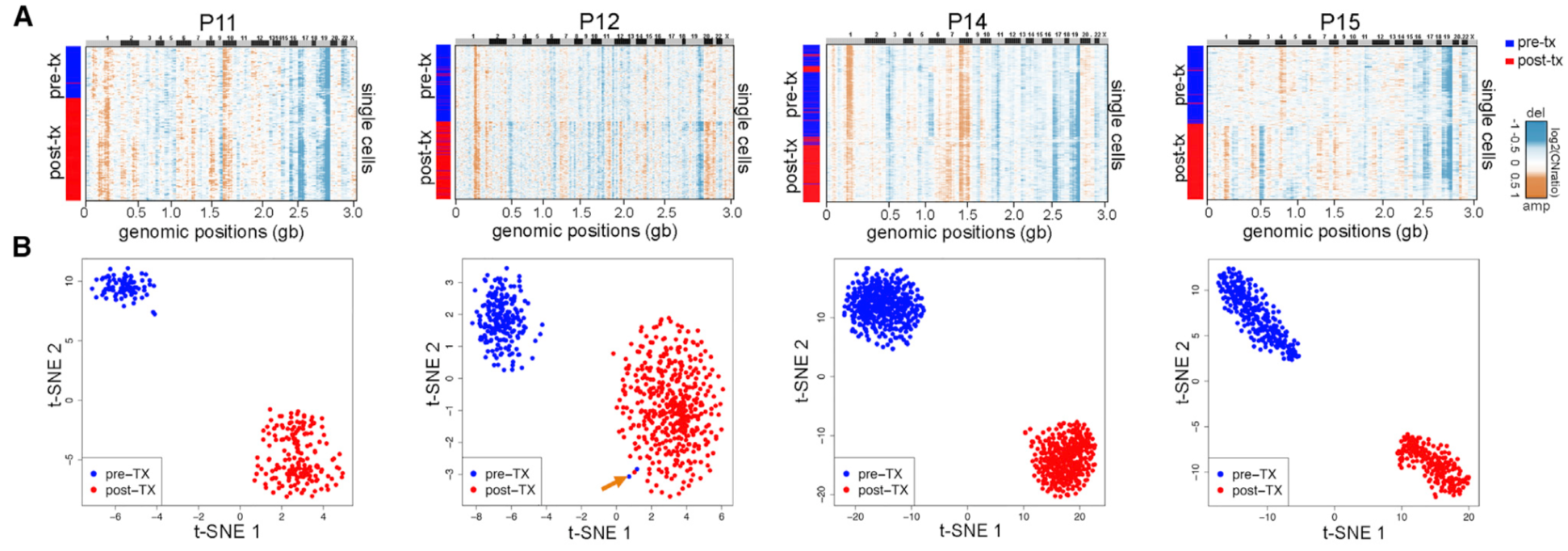
对于那些新辅助化疗抵抗的病人来说,可以看到他们治疗后仍然是癌症细胞,而且是跟治疗前区别很大的癌细胞。
单细胞转录组数据找差异基因
对于那些新辅助化疗受益的病人来说,因为治疗后癌症细胞急剧减少,所以比较正常细胞及癌症细胞即可,可以看到常见的12个癌症相关基因都是显著差异的。
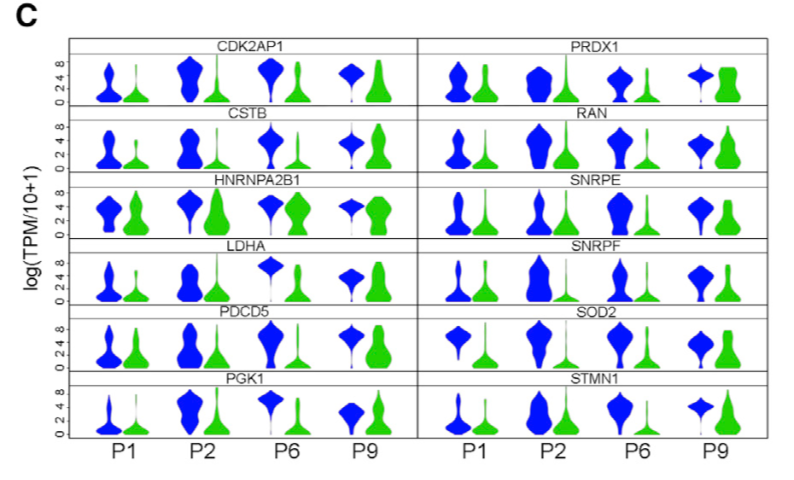
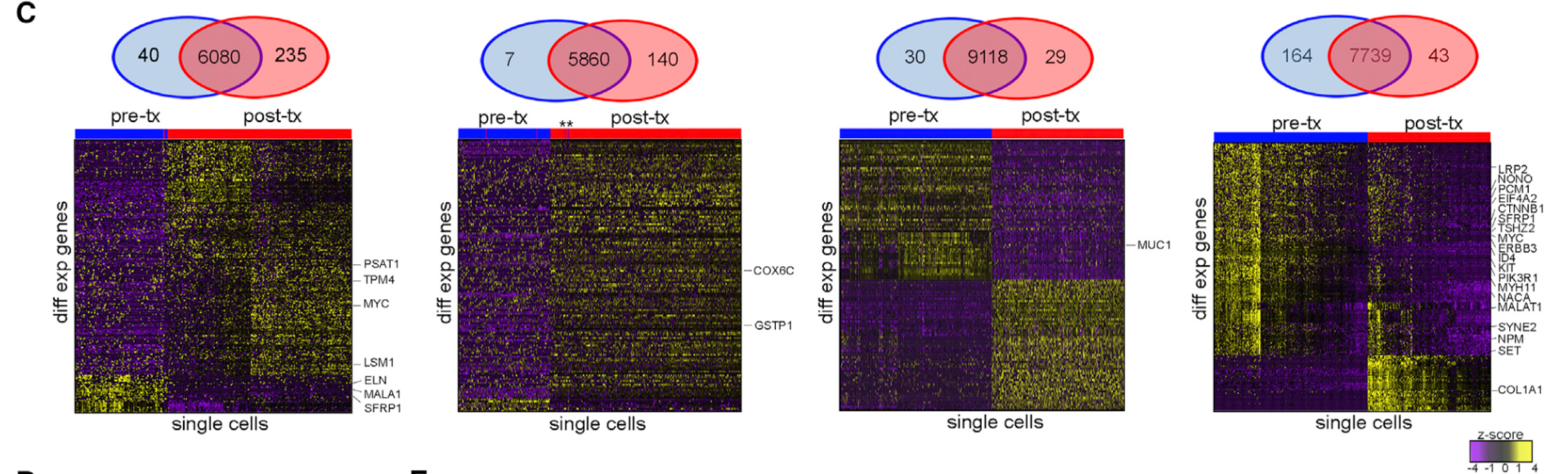
对于那些新辅助化疗抵抗的病人来说,因为治疗前后都存在癌症细胞,所以可以看看它们之间以及它们分别于正常细胞的表达差异基因。
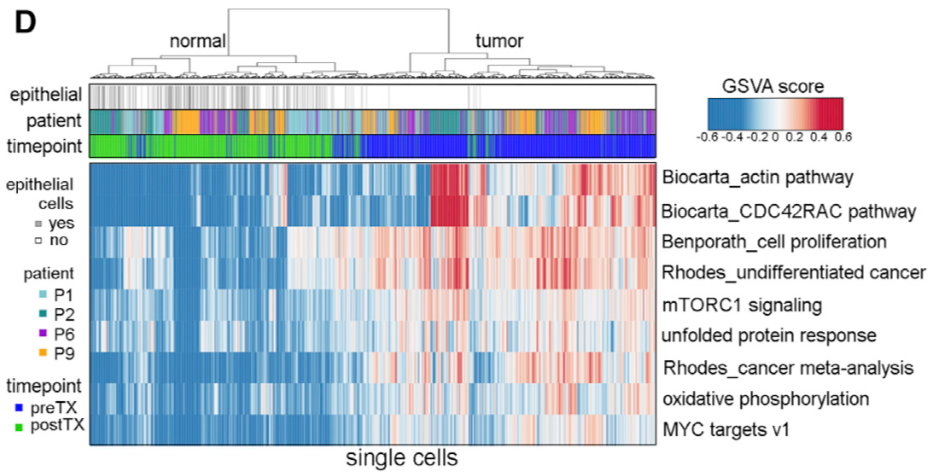
使用GSVA研究各种分子通路
在那些新辅助化疗受益的病人,很明显看到一些癌症相关通路能显著的区分正常细胞和癌症细胞。
在那些新辅助化疗抵抗的病人也可以很明显的看到这些癌症相关分子通路都在治疗前后有信号,虽然有差异,说明了治疗前后病人的肿瘤部分发生了变化。
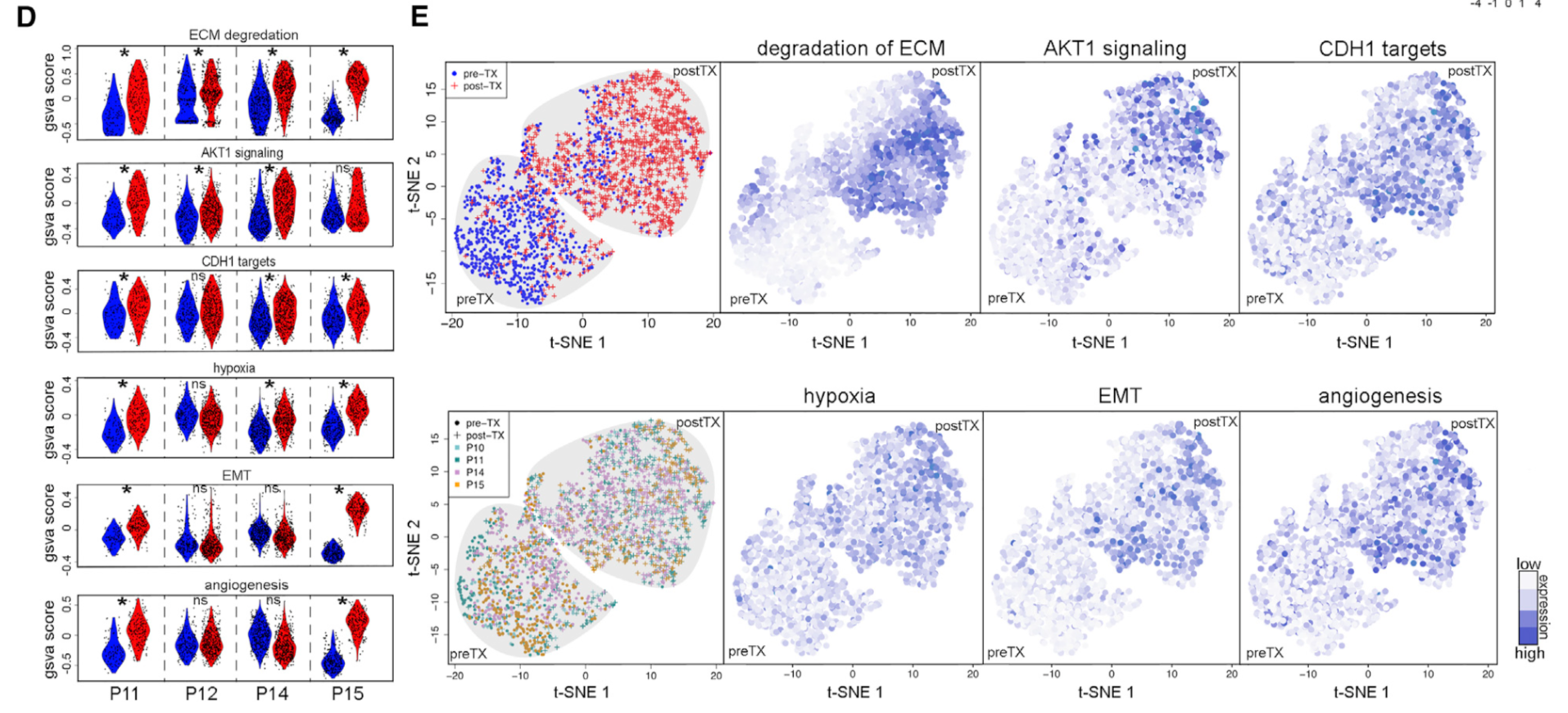
全文精华
首先是精炼了肿瘤治疗进化模型;
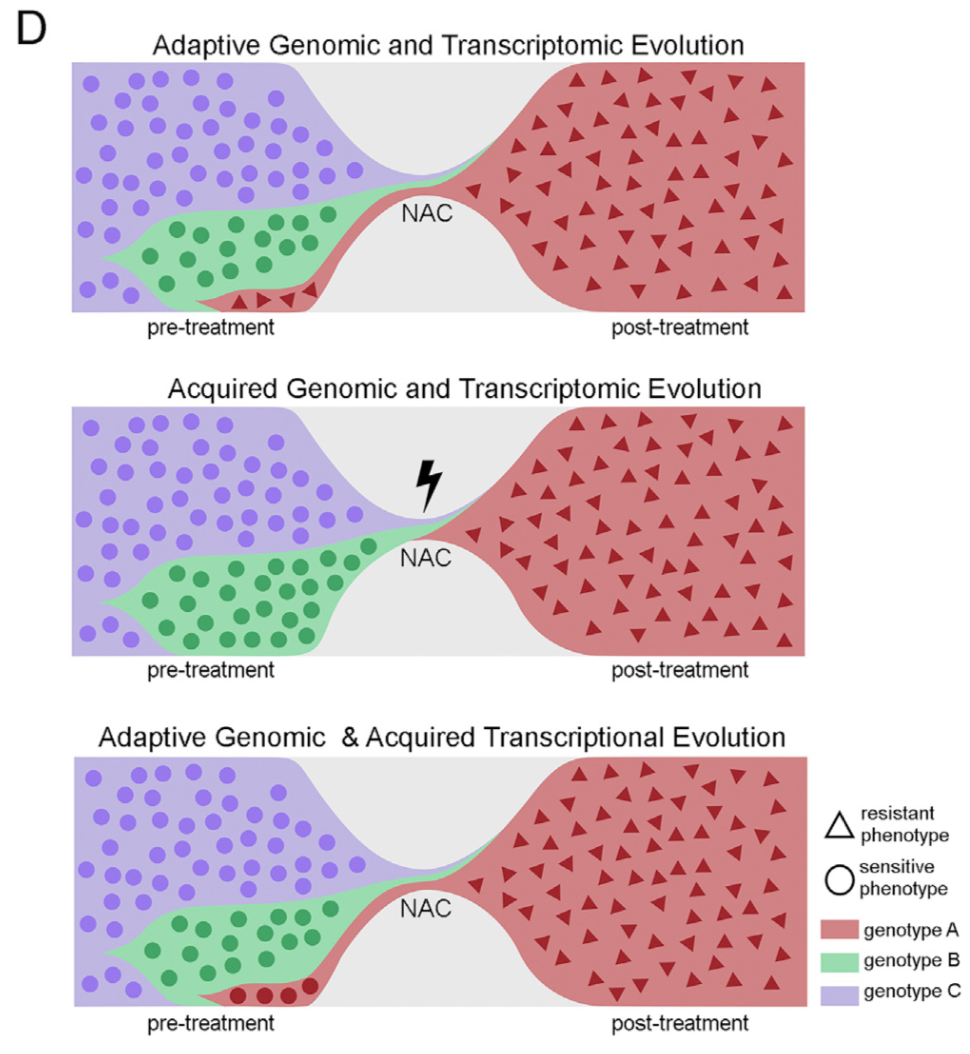
然后是定义了新辅助化疗抵抗的基因集
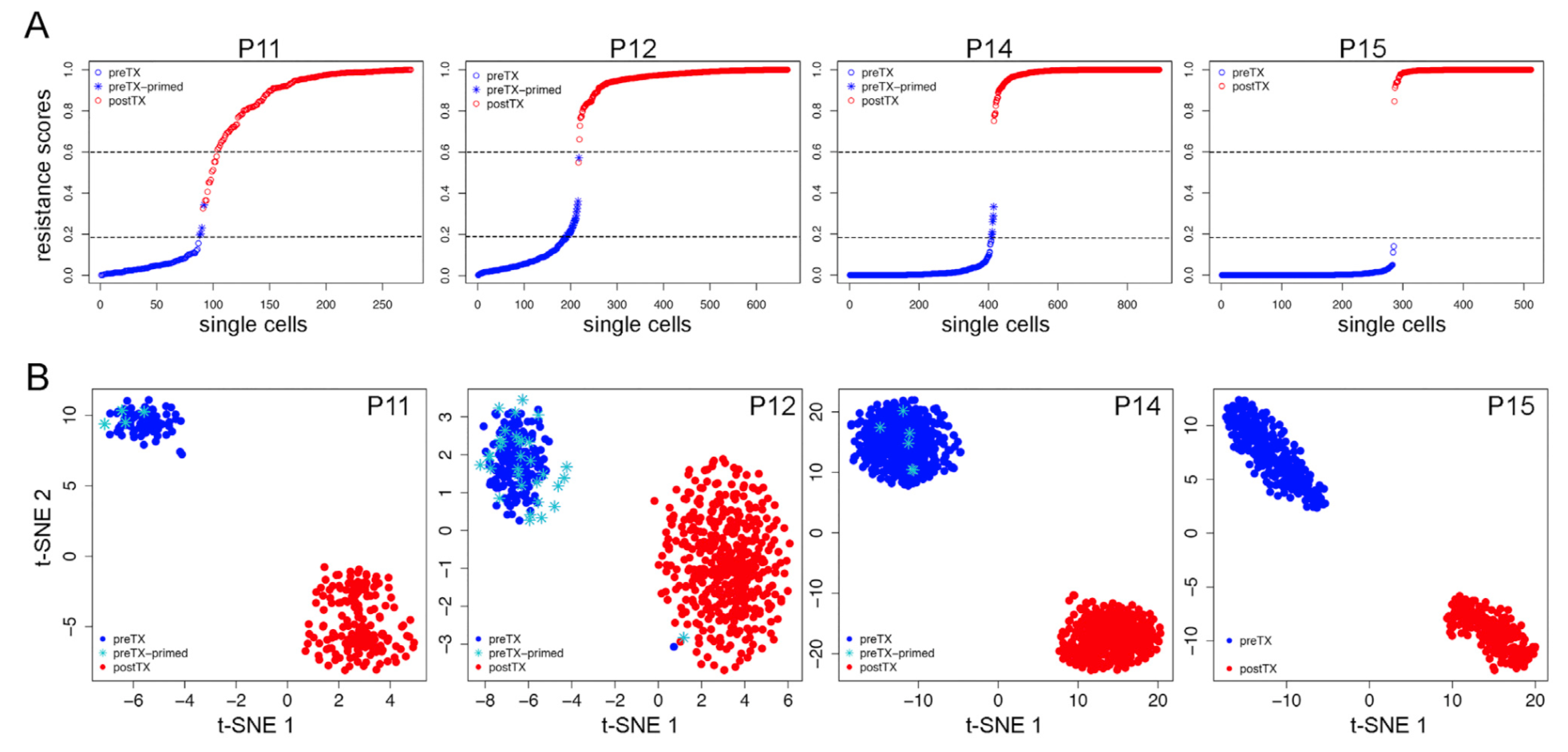
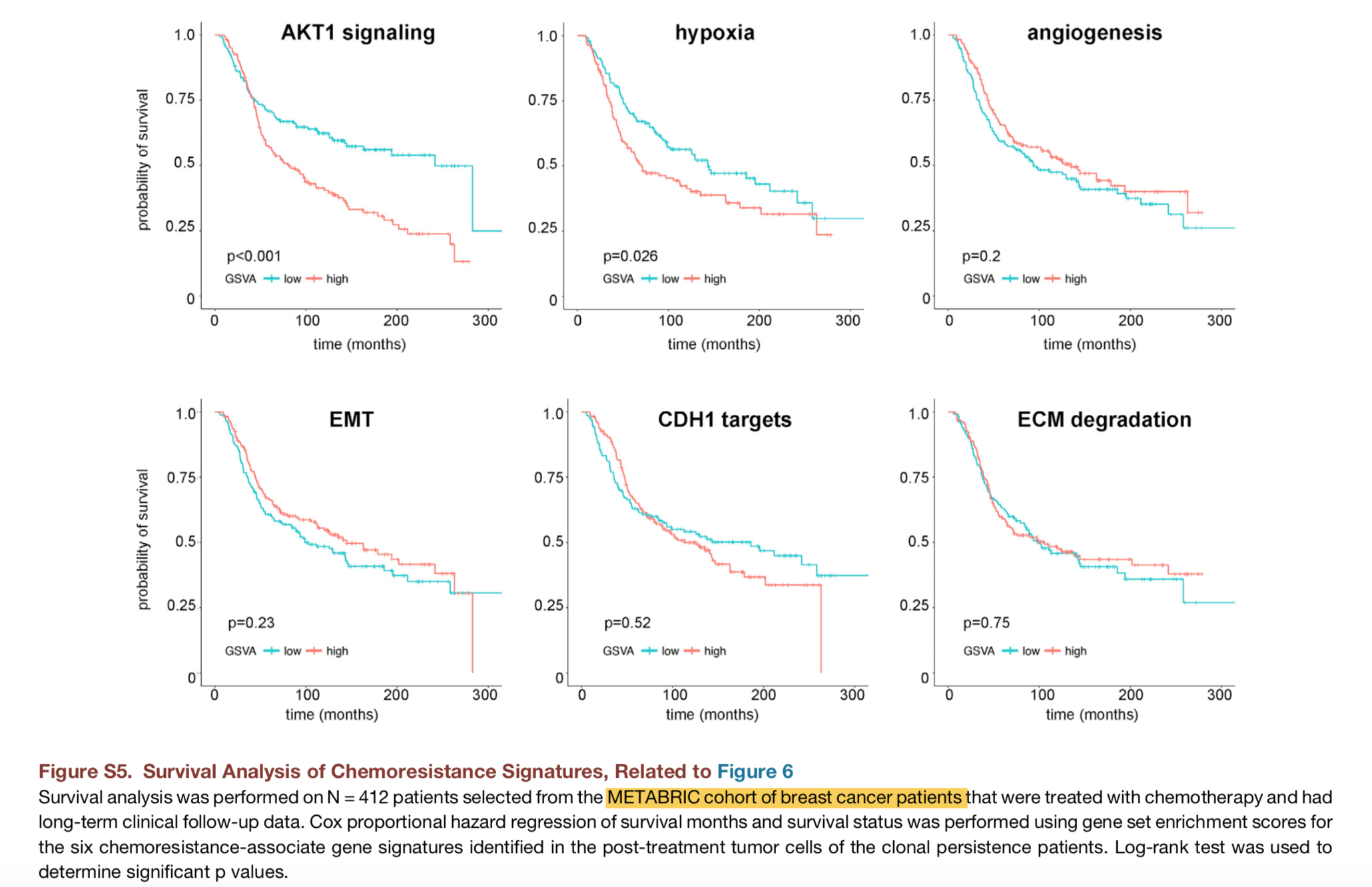
最后的锦上添花
可有可无的分析,比如在METABRIC数据集在做生存分析
以及根据PAM50来对乳腺癌单细胞转录组表达矩阵分类:
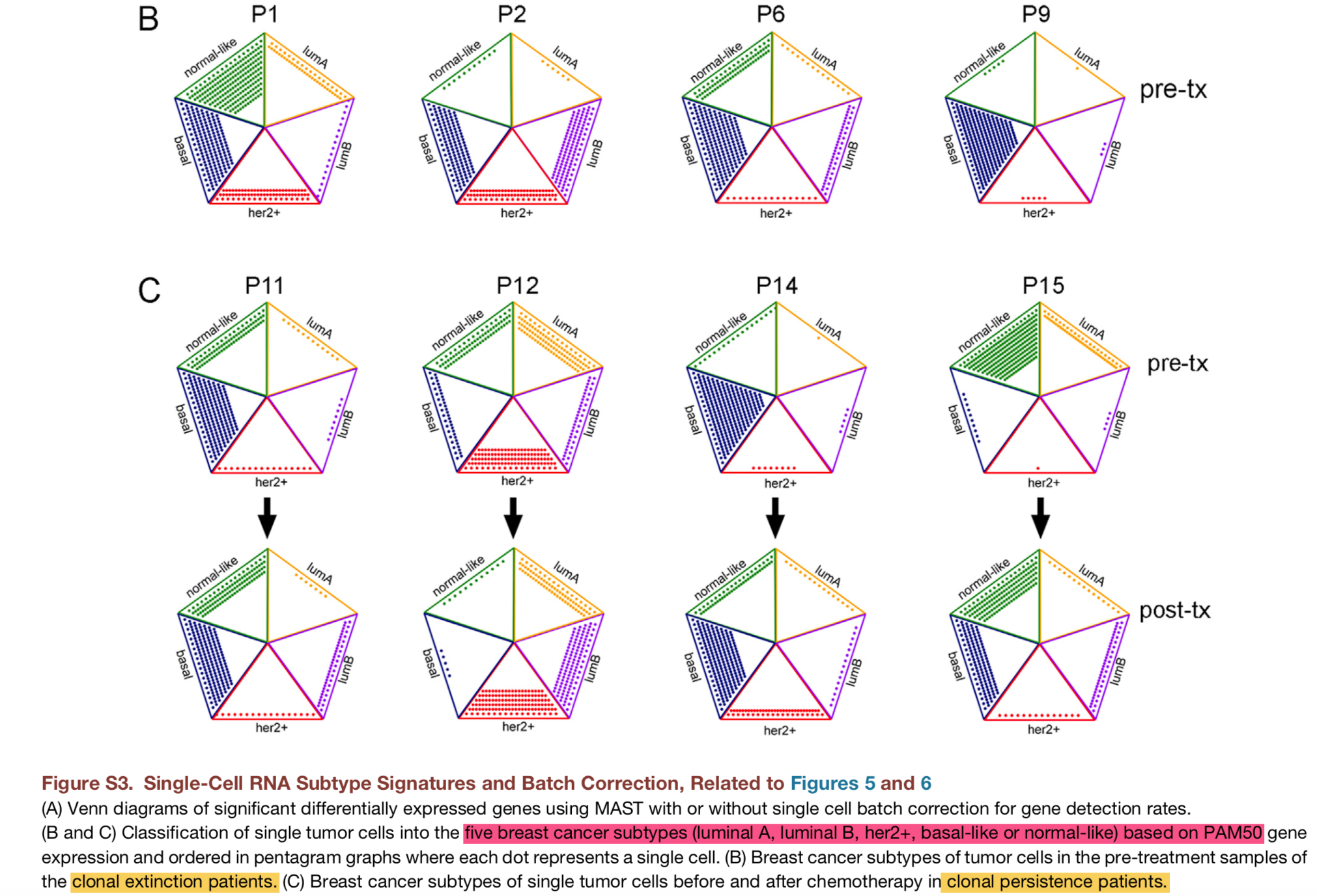
数据在NCBI
可以下载 :PRJNA396019 • SRP114962 • 数据: https://www.ncbi.nlm.nih.gov/Traces/study/?acc=SRP114962
下载metadata可以看到共 4847 TRANSCRIPTOMIC 单细胞转录组和 901 (其中829个单细胞,72个bulk的WES)GENOMIC数据,详情如下:1151 RNA-Seq mid 1487 RNA-Seq post 2209 RNA-Seq pre 275 WGS midTX 183 WGS postTX 371 WGS preTX 11 WXS 2cycleschemo 21 WXS blood 19 WXS operative 21 WXS pre选择4个新辅助化疗受益及4个抵抗的病人做单细胞测序,包括
- single-cell DNA sequencing to analyze 900 cells
- single-cell RNA sequencing to analyze 6,862 cells.