2018年9月朋友圈刷屏的nature文章,题目是:Accurate classification of BRCA1 variants with saturation genome editing 字面意思是探索了BRCA1基因上面的全部可能的突变位点的生物学意义,但事实上只是探索了96.5% of all possible single-nucleotide variants (SNVs) in 13 exons that encode functionally critical domains of BRCA1.
具体数目如下:
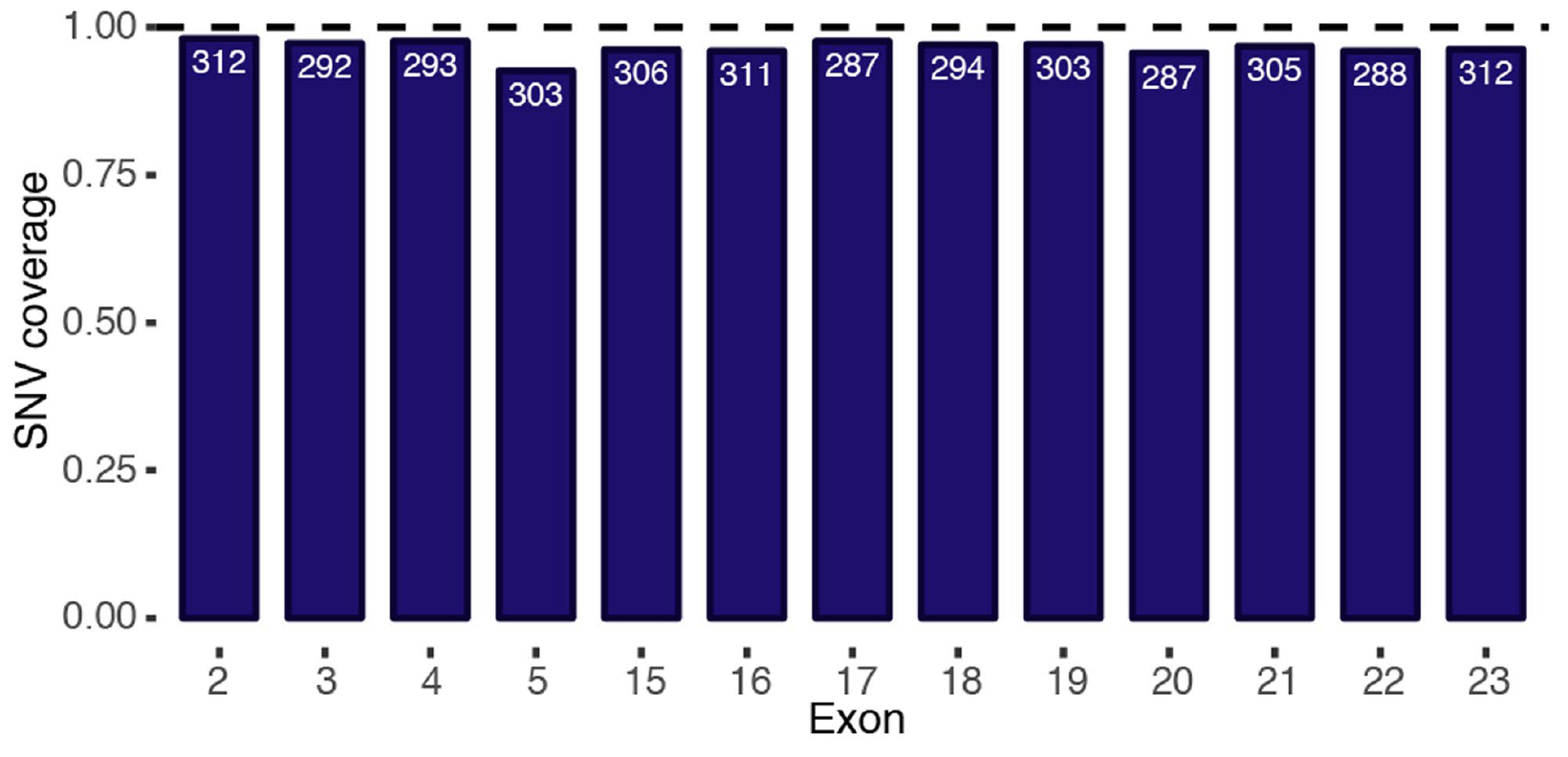
不过这个方法的确是很大的创新,只不过工作量太大,如果想真正的全基因组全部位点都探索一遍。
背景知识
BRCA1基因
BRCA1是一种人体肿瘤抑制基因,BRCA1失去功能的突变与易患早发型乳腺癌和卵巢癌有关。虽然已发现的BRCA1基因变异有数千种,但许多都被列为“意义未明的变异”,给患癌风险评估带来了巨大挑战。
variants of uncertain significance (VUS)
基因突变源于细胞分裂,但是并不是所有的变异都与疾病有关,它们被分成“良性”、“可能良性”“不确定重要性的变异”、“可能致病”和“致病性的”。
意义不明确的突变(即VUS),包括基因与以及发表的DNA基因有着不同序列的基因,并且既不能归入有害突变的类别、也不能归入无害突变的类别。
VUS报告解读的难点首先就是VUS非常难以分类。这是因为:
- 首先,是目前并无一致的报告内容和格式:有的报告了变异但是并无解释、有的使用了局部地区的指南。
- 其次,是我们需要更多的证据来帮助我们确定某些突变确实是与癌症相关的。
目前有2种实验方案来评价一个VUS,但是都太低效。细胞系HAP1
特殊的细胞系HAP1。在这种细胞系里,一旦BRCA1蛋白失去其应有的功能,细胞就会死亡。 基于此,美国华盛顿大学的Jay Shendure及同事运用基因组编辑技术,对BRCA1基因功能至关重要的13个外显子上的近4000种单核苷酸变异 的功能进行了评估,并在2000万个人类单倍体 HAP1 细胞中进行了后续细胞存活率测定 。
saturation genome editing (SGE)
他们每次在2000万个细胞内,对一整段外显子进行基因编辑。随后,研究人员们先让这些细胞生长11天,再做后续的分析。如果由于某个特定突变导致BRCA1失去其功能,相应的细胞就会死亡,后续分析中也就更难找到这种特定突变的存在。
RNA score
懒得看了。
Function score
作者的计算结果提供登记后的下载:https://sge.gs.washington.edu/BRCA1/ 计算方法如下:
- we first calculated the log2ratio of the frequency of a SNV on day 11 to its frequency in the plasmid library.
- Second, positional biases in editing rates were modelled using day 5 SNV frequencies and subtracted
- Third, to enable comparisons between exons, we normalized function scores such that the median synonymous and nonsense SNV in each experiment matched global medians. Lastly, a small number of SNVs that could not confidently be scored were filtered out
可以简单理解,Function score 越小,代表该SNV对BRCA1基因的功能影响越大,所以细胞死的越快,说明该SNV是有害的,但是在这里作者玩了一个文字游戏。
有害的SNV作者命名为non-functional,其实说的是正是因为这个SNV有害,所以导致了BRCA1基因的non-functional。HAP1细胞系验证BRCA1突变的影响
首先根据Function score 对SNV进行分类,因为这4千个SNV的得分分布很有规律,如下:
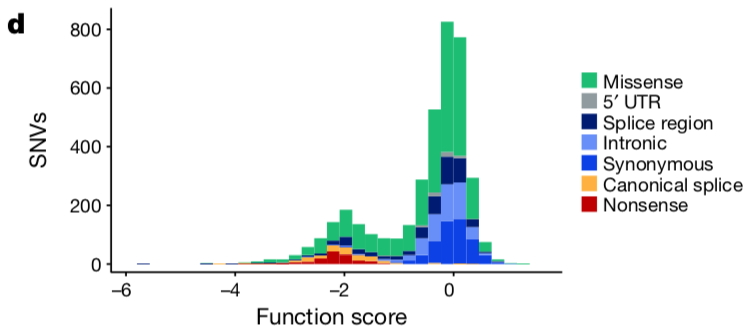
All nonsense SNVs scored below −1.25 (n = 138, median = −2.12), whereas 98.7% of synonymous SNVs more than 3 bp from splice junctions scored above −1.25 (n = 544, median = 0.00).
所以可以使用two-component Gaussian mixture model 来进行分类: - 72.5% of SNVs as functional
- 21.1% as non-functional
- 6.4% as intermediate
值得注意的是SNVs disrupting canonical splice sites (the two intronic positions immediately flanking each exon) were mostly non-functional (89.5%) or intermediate (5.5%) 非常符合预期,因为那些发生在剪切位点的SNV通常我们认为非常有害,所以导致了BRCA1基因的功能缺失。
而那些NVs positioned 1–3 bp into the exon or 3–8 bp into the intron had variable effects. We defined SNVs in these regions that did not alter the amino acid sequence as ‘splice region’ variants, of which 22.9% were non-functional,有害程度不如 canonical splice sites ,所以造成BRCA1基因的功能缺失的比例也不高。
一切都是那么的符合期望!!!临床评估的高度相关性
但是,符合生物学的期望是远远不够的,还需要结合临床资料,如下图:
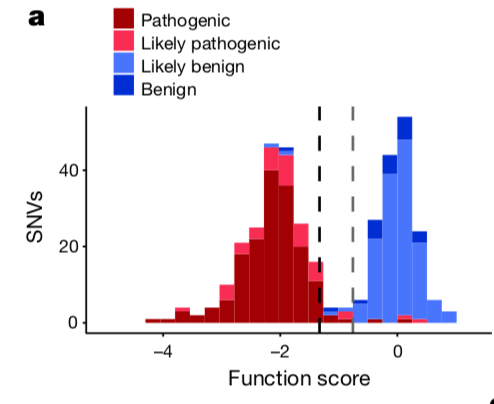
这个就不得了啦,因为跟ClinVar数据库进行了对比,而我们通常认为ClinVar对SNV的分类是专家们达成共识的!
首先 Of 169 SNVs deemed ‘pathogenic’ in ClinVar that overlapped with our classifications, 162 were designated ‘non-functional’, two ‘functional’, and the remaining five ‘intermediate’. 也就是说 专家们认为临床有害突变的那些位点,恰好是作者的SGE筛选到的那些导致BRCA1基因功能没掉了的non-functional的SNV。
同理,of 22 SNVs deemed ‘benign’ in ClinVar, 20 were designated ‘functional’, one ‘non-functional’, and one ‘intermediate’
这个预测模型的AOC和AUC可以说是吊炸天了!!!
正是因为在专家们明确致病还有明确是良性的突变分类上面大展身手,作者这个时候就飘了,可以来攻克那些 意义不明确的突变(即VUS)啦!!!
具体细节不用看了,总之作者认为的系统很牛气!
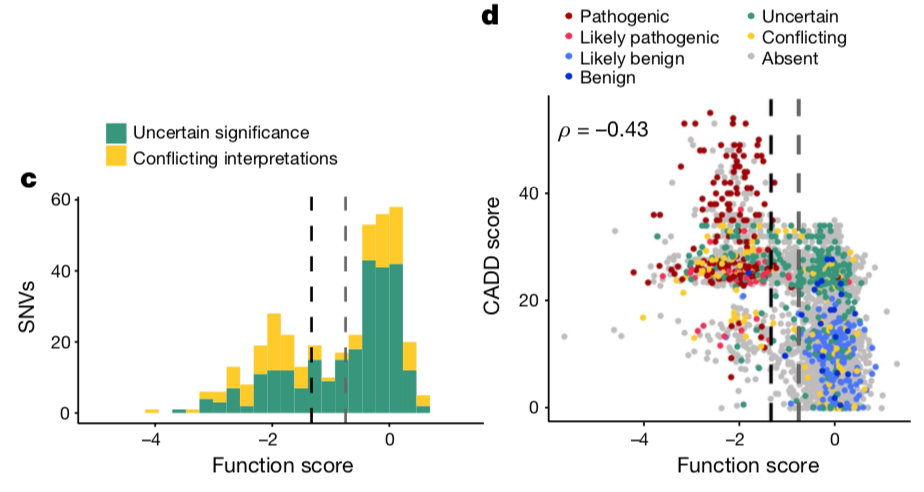
所以作者很自豪的得出结论: 因为在临床数据库的高度一致性,作者肯定了其功能评分对变异分类的重要价值,并认为研究结果可直接应用于BRCA1基因筛查的解读。其它确定VUS的方法
2018年9月25日,《Journal of the American Medical Association》期刊发表了这一篇题为“Prevalence of Variant Reclassification Following Hereditary Cancer Genetic Testing”的文章,作者回顾145万人的遗传性癌症基因检测结果发现,近25%的“不确定重要性的基因突变”随后被重新分类——有些与癌症相关的可能性较低,有些与癌症相关的可能性较高。
典型的基因约包含27000个碱基对(A-T,G-C),这些大量的碱基对意味着无数的潜在变异。随着高通量肿瘤测序和胚系基因检测的广泛应用,如何应对临床意义不明确的基因突变,是大家面临的挑战之一。这项研究回顾了2006年-2016年间Myriad Genetics实验室以及西南医学中心癌症遗传学项目的相关临床数据。总体而言,24.9%的“不确定重要性的变异”被重新分类,其中大多数降级为良性。
这项研究提醒医生需要知道关于基因突变的知识更新有多快,重新分类很常见。同时,独立的商业型实验室需要定期检查基因突变数据,并提醒医生注意重新分类。即便从“不确定”重新分类为“良性”,对于病人来说也很重要,因为可以让他们安心。