也是公共数据的挖掘,但是不是对TCGA数据库的挖掘,不过其整合的多组学又恰好是TCGA计划纳入的7种数据。
文章标题很精炼:Modeling precision treatment of breast cancer. 是2013年发表的,但是在2015有一个勘误信息,修正了补充材料的一些图表,还有GATK的流程。
目前还不清楚不同组学数据该如何结合起来更好的预测癌症病人的治疗效果,考虑到细胞系数据容易获取,也比较方便做实验,所以作者整合了一些细胞系的公共数据。
乳腺癌病人的异质性在治疗领域是很大的问题,有文章根据表达量分6类,如下:
- luminal A
- luminal B
- ERBB2-enriched
- basal-like
- claudin-low
- normal-like
当然,PAM50的分类也是可以的,也有文章把CNV和表达量结合起来把乳腺癌病人分成10类,如果再结合更多的信息,分类会更复杂。这也就是为什么会有TCGA计划,而且即使有了多组学数据,目前也不清楚那一类型的数据更优。
所以本文作者使用2种机器学习算法来针对多组学数据结果进行预测乳腺癌细胞系的药物疗效。
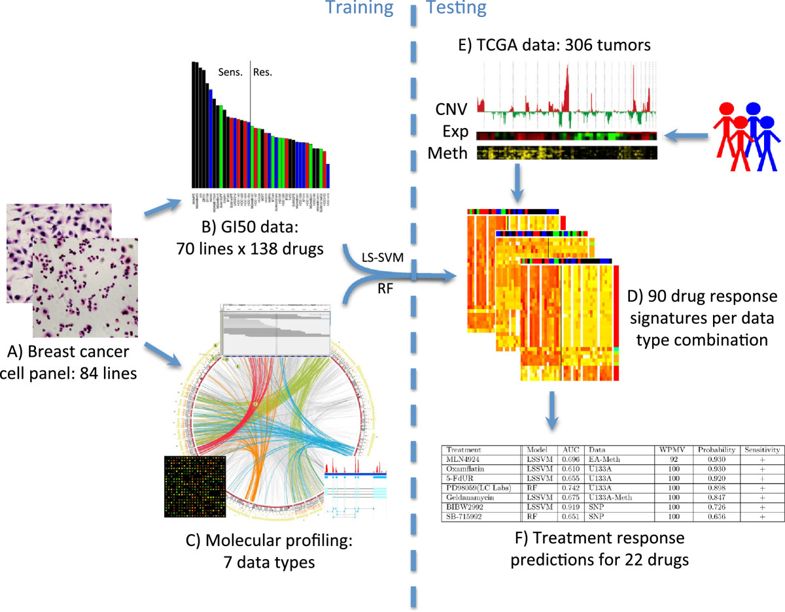
最后还把预测结果应用到了TCGA计划的306个乳腺癌病人数据里面。项目设计
总共是 84 breast cancer cell lines ,包括
- 35 luminal
- 27 basal
- 10 claudin-low
- 7 normal-like
- 2 matched normal
- 3 of unknown subtype.
本研究纳入了7种不同类型的数据: - DNA copy number (Affymetrix SNP6 - EGA accessions EGAS00000000059 and EGAS00001000585)
- mRNA expression (Affymetrix U133A and Exon 1.0 ST array - ArrayExpress accessions E-TABM-157 and E-MTAB-181)
- transcriptome sequence (RNAseq - Gene Expression Omnibus (GEO) accession GSE48216)
- promoter methylation (Illumina Methylation27 BeadChip - GEO accession GSE42944)
- protein abundance (Reverse Protein Lysate Array - Additional file 2)
- mutation status (Exome-Seq - GEO accession GSE48216).
- therapeutic response data
每种数据的样本量是: - Exome-seq data were available for 75 cell lines
- SNP6 data for 74 cell lines
- therapeutic response data for 70
- RNAseq for 56
- exon array for 56
- Reverse Phase Protein Array (RPPA) for 49
- methylation for 47
- U133A expression array data for 46 cell lines.
只有48个细胞系有着4种以上的数据。
整体项目设计如下:
不同数据的相关性: - Correlation among the three expression datasets (U133A, exon array, and RNAseq) ranged from 0.6 to 0.77 at the cell line level, and from 0.58 to 0.71 at the gene level.
- Promoter methylation and gene expression were, on average, negatively correlated as expected, with correlation ranging from -0.16 to -0.25 at the cell line level and -0.10 to -0.15 at the gene level.
- Across the genome, copy number and gene expression were positively correlated (0.18 to 0.22 at the cell line level; 0.35 to 0.44 at the gene level).
这样的简单粗暴的比较并不可取。预测算法
使用了2种机器学习算法:
- weighted least squares support vector machine (LS-SVM)
- random forests (RF)
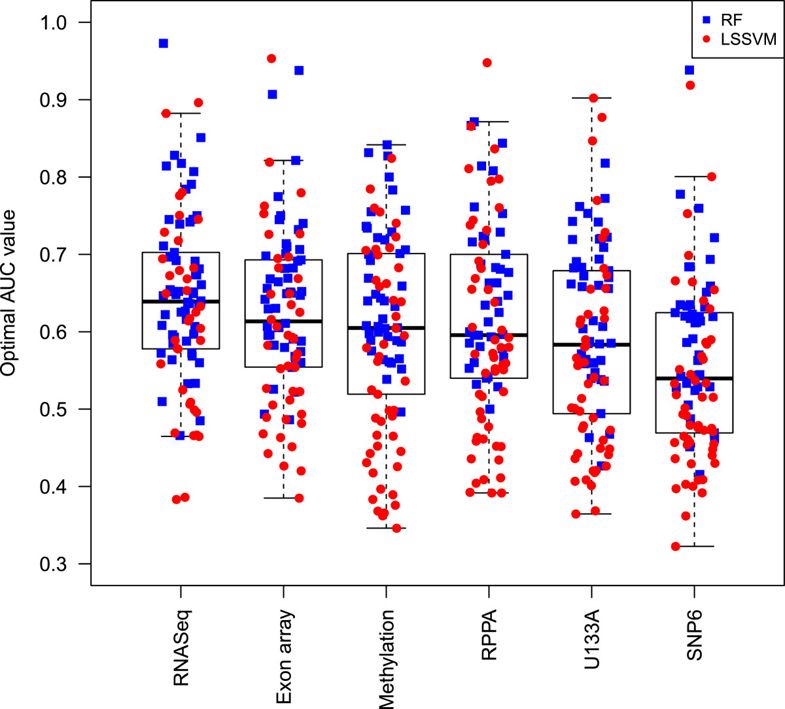
每个算法在不同数据类型的有不同的效果比如,对:LS-SVM classifiers - RNAseq performed best for 22 compounds
- exon array for 20 compounds
- SNP6 for 18,
- U133A for 17
- methylation data for 12 compounds
而对RF算法,表现差不多,总体来说,RNAseq表现最好,而SNP6表现最差,如下图:
meta分析验证signatures
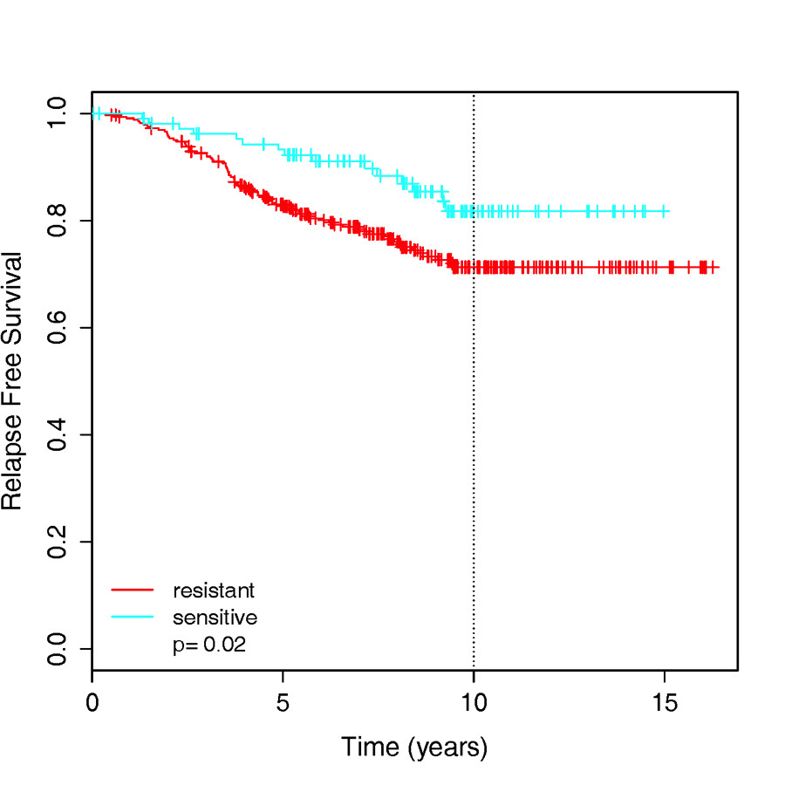
作者这里针对tamoxifen药物得到了174-gene signature可以用来区分药物敏感性和耐受性,然后就整合了4篇文献里面的439 ER-positive patients把他们分组后根据 relapse-free survival 信息来做生存分析,如下:
数据文章来源是:
- Definition of clinically distinct molecular subtypes in estrogen receptor-positive breast carcinomas through genomic grade. J Clin Oncol. 2007
- The 76-gene signature defines high-risk patients that benefit from adjuvant tamoxifen therapy. Breast Cancer Res Treat. 2009
- Genomic index of sensitivity to endocrine therapy for breast cancer. J Clin Oncol. 2010
- Gene expression profiling in breast cancer: understanding the molecular basis of histologic grade to improve prognosis. J Natl Cancer Inst. 2006
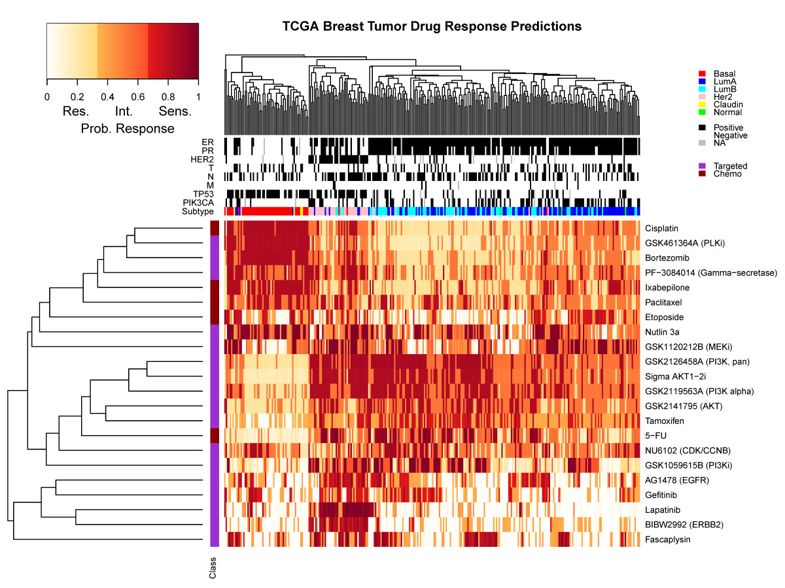
TCGA数据库验证
最后还在306 TCGA breast tumors for which expression (Exp), copy number (CNV) and methylation (Meth) measurements 数据里面进行验证,预测为 resistant, intermediate or sensitive 的分组。