这个需求实在是太常见了,很多时候我们下载的表达矩阵,都是基因的探针ID作为行名来标记,如下:

这个变量是 dat,一个表达矩阵, 所以需要对探针进行注释,并且筛选。
首先看看注释的结果:
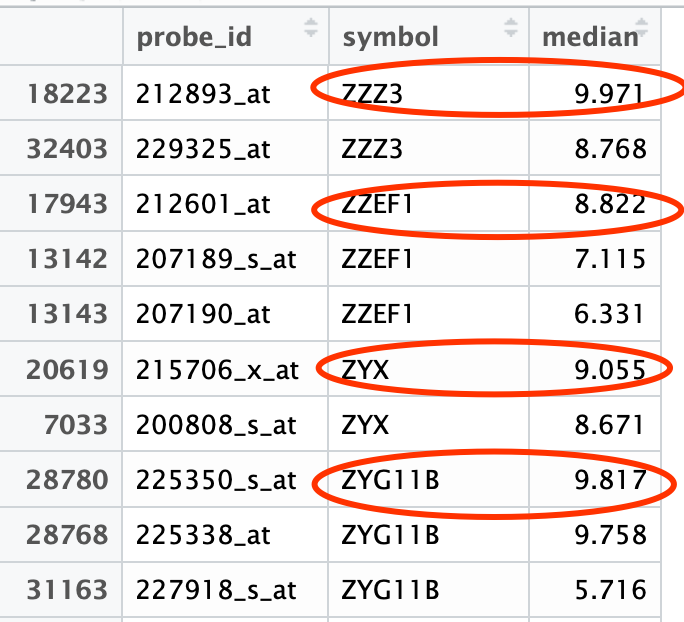
很明显可以看到,很多基因都是有多个探针的,而且每个探针的中位表达量不一样,这里我们可以选择最大的探针保留。
代码很简单:
dat[1:4,1:4]
library(hgu133plus2.db)
ids=toTable(hgu133plus2SYMBOL)
head(ids)
dat=dat[ids$probe_id,]
dat[1:4,1:4]
ids$median=apply(dat,1,median)
ids=ids[order(ids$symbol,ids$median,decreasing = T),]
ids=ids[!duplicated(ids$symbol),]
dat=dat[ids$probe_id,]
rownames(dat)=ids$symbol
dat[1:4,1:4]
过滤后的表达矩阵如下:
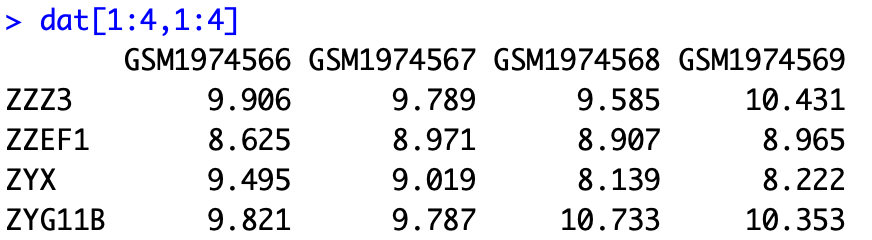
可以手动简单检查一些探针的转换情况来验证代码是否准确无误。
不过,如果你连表达矩阵都不知道如何生成,那么你需要补充的基础知识就比较多了。
目录:
解读GEO数据存放规律及下载,一文就够
解读SRA数据库规律一文就够
从GEO数据库下载得到表达矩阵 一文就够
GSEA分析一文就够(单机版+R语言版)
根据分组信息做差异分析- 这个一文不够的
差异分析得到的结果注释一文就够