PS: 请不要在问我关于这个包的任何问题,直接联系Y叔,我就两年前用过一次而已,再也没有用过。
Y叔的包更新太频繁了,这个教程已经作废,请不要再照抄了,可以去我们论坛看新的教程:http://www.biotrainee.com/thread-1084-1-1.html
一:下载安装该R包
clusterProfiler是业界很出名的YGC写的R包,非常通俗易懂,也很好用,可以直接根据cuffdiff等找差异的软件找出的差异基因entrez ID号直接做好富集的所有内容;
Bioconductor网站上面有关于它的介绍,按照上面说的方式来安装即可
http://www.bioconductor.org/packages/release/bioc/html/clusterProfiler.html
source("http://bioconductor.org/biocLite.R");biocLite("clusterProfiler")
二、输入数据
diff_gene.entrez文件,是通过各种差异基因软件找出来的差异基因的entrez ID号列表,每一个ID号一行,几百个差异基因就几百行
三、R语言代码
setwd("C:\\Users\\Administrator\\Desktop\\ref")
a=read.table("diff_gene.entrez")
require(DOSE)
require(clusterProfiler)
gene=as.character(a[,1])
ego <- enrichGO(gene=gene,organism="human",ont="CC",pvalueCutoff=0.01,readable=TRUE)
ekk <- enrichKEGG(gene=gene,organism="human",pvalueCutoff=0.01,readable=TRUE)
write.csv(summary(ekk),"KEGG-enrich.csv",row.names =F)
write.csv(summary(ego),"GO-enrich.csv",row.names =F)
四、输出文件解读
看得懂R语言的都知道,这个代码输出了两个文件KEGG-enrich.csv和GO-enrich.csv,就是我们的GO和KEGG富集的结果,其中内容如下
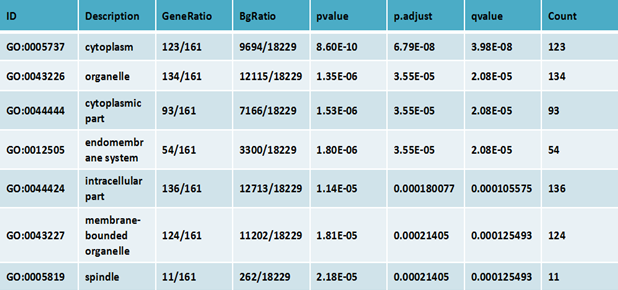
上述表格为差异基因的Gene Ontology富集分析结果表格。
GO ID: Gene Ontology数据库中唯一的标号信息
Description :Gene Ontology功能的描述信息
GeneRatio:差异基因中与该Term相关的基因数与整个差异基因总数的比值
BgRation:所有( bg)基因中与该Term相关的基因数与所有( bg)基因的比值
pvalue: 富集分析统计学显著水平,一般情况下, P-value < 0.05 该功能为富集项
p.adjust 矫正后的P-Value
qvalue:对p值进行统计学检验的q值
Count:差异基因中与该Term相关的基因数
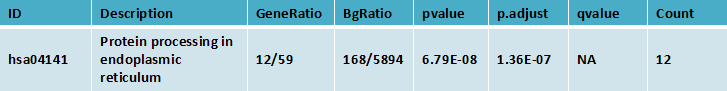
上述表格为差异基因的KEGG Pathway富集分析结果表格。
ID: KEGG 数据库中通路唯一的编号信息。
Description :Gene Ontology功能的描述信息
GeneRatio:差异基因中与该Term相关的基因数与整个差异基因总数的比值
BgRation:所有( bg)基因中与该ID相关的基因数与所有( bg)基因的比值
pvalue: 富集分析统计学显著水平,一般情况下, P-value < 0.05 该功能为富集项
p.adjust 矫正后的P-Value
qvalue:对p值进行统计学检验的q值
Count:差异基因中与该Term相关的基因数
业界不出名的YGC
呀,可是我听说过的就是你呀!
clusterProfiler还真的是好方便啊,支持
如果你真正懂了enrichment,其实你自己都可以写一个这个类似的包