因为昨天看到了TxDb.Hsapiens.UCSC.hg38.knownGene 包来获取基因的坐标及长度跟其它主流数据库有差异,所以今天彻底比较一下TxDb.Hsapiens.UCSC.hg38.knownGene 包和CCDS数据库的差异。
详见:https://mp.weixin.qq.com/s/2QJvQxVECcxpJIsId1pHYA
通过CCDS基因的外显子长度之和
这里我GitHub项目:https://github.com/jmzeng1314/scRNA_smart_seq2/blob/master/RNA-seq/step7-counts2rpkm.R 里面探索过3种方法获取基因长度,然后发现 同样的基因在不同数据库记录的位置信息差距好离谱 所以不得不弃用 TxDb.Hsapiens.UCSC.hg38.knownGene 包。
这里还是使用CCDS记录文件吧,在数据库:ftp://ftp.ncbi.nlm.nih.gov/pub/CCDS/
wget ftp://ftp.ncbi.nlm.nih.gov/pub/CCDS/current_human/CCDS.20180614.txt
cat CCDS.20180614.txt |perl -alne '{/\[(.*?)\]/;next unless $1;$gene=$F[2];$exons=$1;$exons=~s/\s//g;$exons=~s/-/\t/g;print "$F[0]\t$_\t$gene" foreach split/,/,$exons;}'|sort -u |bedtools sort -i >exon_probe.hg38.gene.bed
cat exon_probe.hg38.gene.bed|perl -alne '{$s+=$F[2]-$F[1]}END{print $s}'
## 计算得到 WES 全长是 36540331, 约 38Mb,所以就采用这个吧
cat exon_probe.hg38.gene.bed|perl -alne '{$s{$F[3]}+=$F[2]-$F[1]}END{print "$_\t$s{$_}" foreach keys %s}' > gene_length.human.txt
通过TxDb.Hsapiens.UCSC.hg38.knownGene 包得到
我GitHub项目:https://github.com/jmzeng1314/scRNA_smart_seq2/blob/master/RNA-seq/step7-counts2rpkm.R 里面的第一个代码:
# 参考我GitHub项目:https://github.com/jmzeng1314/scRNA_smart_seq2/blob/master/RNA-seq/step7-counts2rpkm.R
# 获取基因长度。
library(TxDb.Hsapiens.UCSC.hg38.knownGene)
txdb <- TxDb.Hsapiens.UCSC.hg38.knownGene
## 下面是定义基因长度为 非冗余exon长度之和, 即WES的基因长度
if(F){
exon_txdb=exons(txdb)
genes_txdb=genes(txdb)
genes_txdb
?GRanges
# 因为有些基因之间有overlap,所以这个并不是最标准答案。
o = findOverlaps(exon_txdb,genes_txdb)
o
## exon - 1 : chr1 4807893-4807982
## 1 6523
# genes_txdb[6523] # chr1 4807893-4846735 , 18777
t1=exon_txdb[queryHits(o)]
t2=genes_txdb[subjectHits(o)]
t1=as.data.frame(t1)
t1$geneid=mcols(t2)[,1]
# 如果觉得速度不够,就参考R语言实现并行计算
# http://www.bio-info-trainee.com/956.html
#lapply : 遍历列表向量内的每个元素,并且使用指定函数来对其元素进行处理。返回列表向量。
#函数split()可以按照分组因子,把向量,矩阵和数据框进行适当的分组;
#它的返回值是一个列表,代表分组变量每个水平的观测。
g_l = lapply(split(t1,t1$geneid),function(x){
# x=split(t1,t1$geneid)[[1]]
head(x)
tmp=apply(x,1,function(y){
y[2]:y[3]
})
length(unique(unlist(tmp)))
# sum(x[,4])
})
head(g_l)
g_l=data.frame(gene_id=names(g_l),length=as.numeric(g_l))
head(g_l)
save(g_l,file = 'gene_length_of_hg38.Rdata')
}
load(file = 'gene_length_of_hg38.Rdata')
sum(g_l$length)
接下来比较两种数据库的区别
因为两个数据来源的基因长度都计算得到了,就可以在R里面看看它们的相关性:
gl=read.table('gene_length.human.txt')
head(gl)
colnames(gl)=c('symbol','length_CCDS')
load(file = 'gene_length_of_hg38.Rdata')
head(g_l)
colnames(g_l)=c('gene_id', 'length_R')
library(org.Hs.eg.db)
columns(org.Hs.eg.db)
e2s=toTable(org.Hs.egSYMBOL)
head(e2s)
g_l=merge(g_l,e2s,by='gene_id')
comp=merge(g_l,gl,by='symbol')
comp[,3]=log(comp[,3])
comp[,4]=log(comp[,4])
plot(comp[,3:4])
head(comp)
library(ggpubr)
ggscatter(comp,'length_R','length_CCDS')
通过google搜索,重新绘制图代码如下:
library(ggpubr)
p=ggscatter(comp,'length_R','length_CCDS',
color = "black", shape = 21, size = 0.3, # Points color, shape and size
add = "reg.line", # Add regressin line
add.params = list(color = "blue", fill = "lightgray"), # Customize reg. line
conf.int = TRUE, # Add confidence interval
cor.coef = TRUE, # Add correlation coefficient. see ?stat_cor
cor.coeff.args = list(method = "pearson", label.x = 3, label.sep = "\n")
)
p+geom_abline(intercept = 0, slope = 1, color="red",
linetype="dashed", size=1.5)
效果如下:
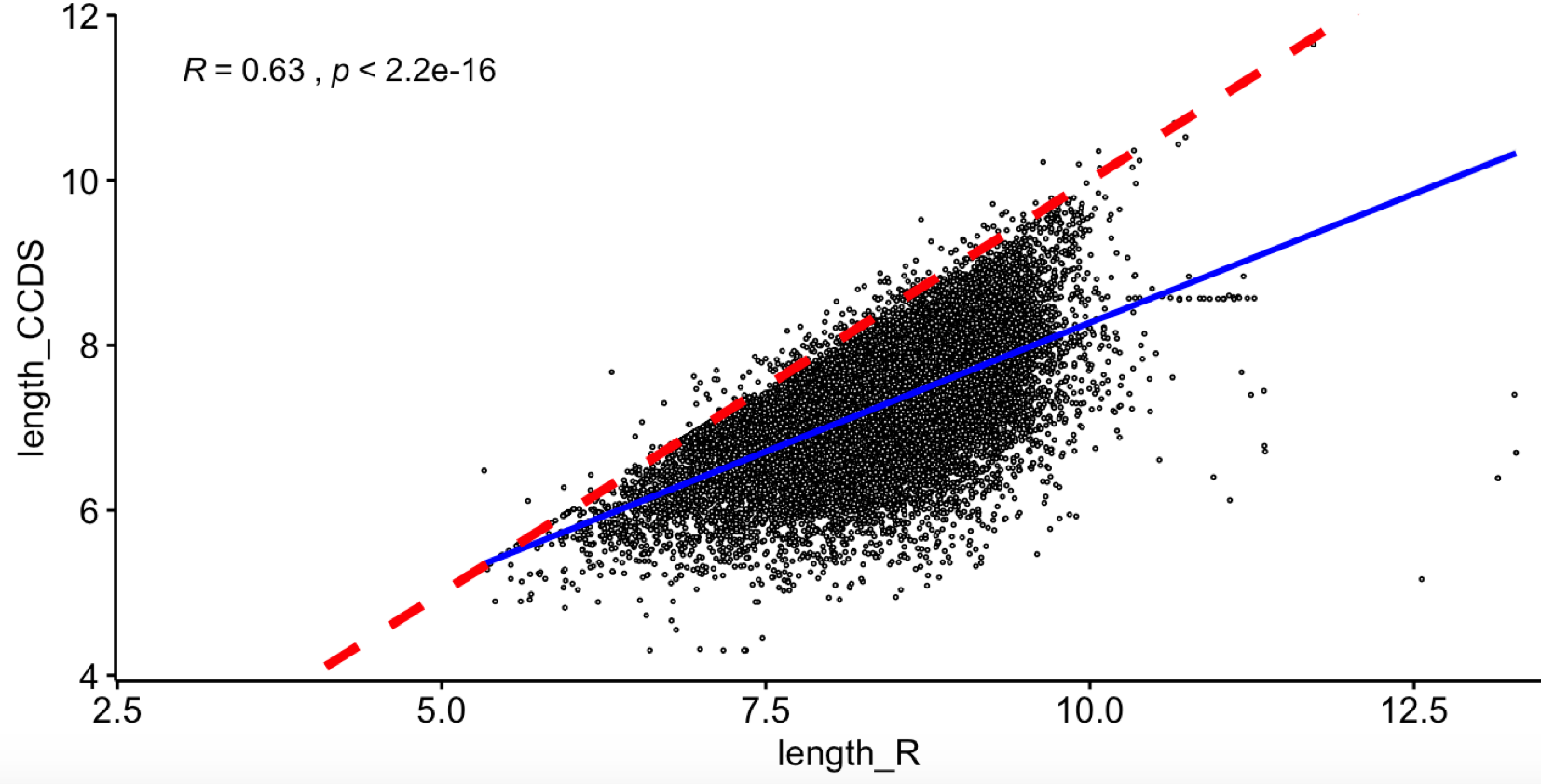
可以很明显看到,通过TxDb.Hsapiens.UCSC.hg38.knownGene 包选择非冗余外显子坐标之后作为基因长度,是会有搞大基长度的倾向,这个是无法避免的, 因为CCDS数据库记录的是基因的CDS,而不是外显子之后,前面我们就强调过,5端和3端的外显子是UTR区域,不是CDS。
然后值得注意的是那些离群点,就是目前主流数据库维护者都束手无策的基因,它们的基因组定位很模糊,我们人类对它们的研究很有限。
我们再看看最长转录本长度
library(TxDb.Hsapiens.UCSC.hg38.knownGene)
txdb = TxDb.Hsapiens.UCSC.hg38.knownGene
tx_by_gene = transcriptsBy(txdb, by="gene")
gene_lens = as.data.frame(max(width(tx_by_gene)))
gene_lens$gene_id=rownames(gene_lens)
colnames(gene_lens)=c('length_R','gene_id')
# 然后走上面同样的流程,发现图也并没有太大的区别,只不过是我昨天拿来做特例的基因的离谱程度被削弱了