首先要了解什么是肿瘤异质性:http://cnvkit.readthedocs.io/en/latest/heterogeneity.html
肿瘤异质性可分为肿瘤间异质性和肿瘤内异质性,其中肿瘤内异质性 Intra-tumor heterogeneity (ITH) 指的是,随着癌细胞的不断生长,其分裂后的子代细胞呈现出与同代细胞或者父细胞的不同,从而使得其各个方面有了较大的差异。
此教程的主角是:PyClone, 那它可以做什么呢,简单的说,就是对存在异质性的肿瘤来说,多次不同部位取样,或者不同时间取样,那么虽然说是同一个肿瘤病人,他们不同的测序结果理论上不一致的, 那么pyclone就可以分析出来,同一个病人的不同测序数据里面共有哪些亚克隆。
可以从GitHub主页上面下载该软件:https://github.com/aroth85/pyclone
## based on 2.7
conda create --name pyclone python=2
source activate pyclone
conda install -c aroth85 pyclone
mkdir ~/biosoft/pyclone
cd ~/biosoft/pyclone
git clone https://github.com/aroth85/pyclone
cd ~/biosoft/pyclone/pyclone/examples/mixing/tsv/
运行测试文件
进入 examples文件夹 , 可以看到测试数据:
-rw-r--r-- 1 jmzeng staff 802B Mar 12 09:22 README.txt
-rw-r--r-- 1 jmzeng staff 9.6K Mar 12 09:22 SRR385938.tsv
-rw-r--r-- 1 jmzeng staff 10K Mar 12 09:22 SRR385939.tsv
-rw-r--r-- 1 jmzeng staff 9.9K Mar 12 09:22 SRR385940.tsv
-rw-r--r-- 1 jmzeng staff 10K Mar 12 09:22 SRR385941.tsv
上面的4个数据作为输入文件,跑主流程如下:
PyClone run_analysis_pipeline --in_files SRR385938.tsv SRR385939.tsv SRR385940.tsv SRR385941.tsv --working_dir pyclone_analysis
在pyclone_analysis文件下会生成如下文件夹或文件
config.yaml #指定用于PyClone分析的设置文件
plots/ #包括生成的全部图
tables/ #包括生成的全部表格
trace/ #包括MCMC抽样算法的原始痕迹
yaml/ #存放yaml突变文件的文件夹,用于PyClone分析
值得注意的是,虽然上面是4个数据文件,但其实可以看做是同一个人的4个数据,所以理论上pyclone是针对单独的一个病人来进行计算,一个病人可以有多个肿瘤部位进行测序,这样pyclone就能计算它们的进化情况。
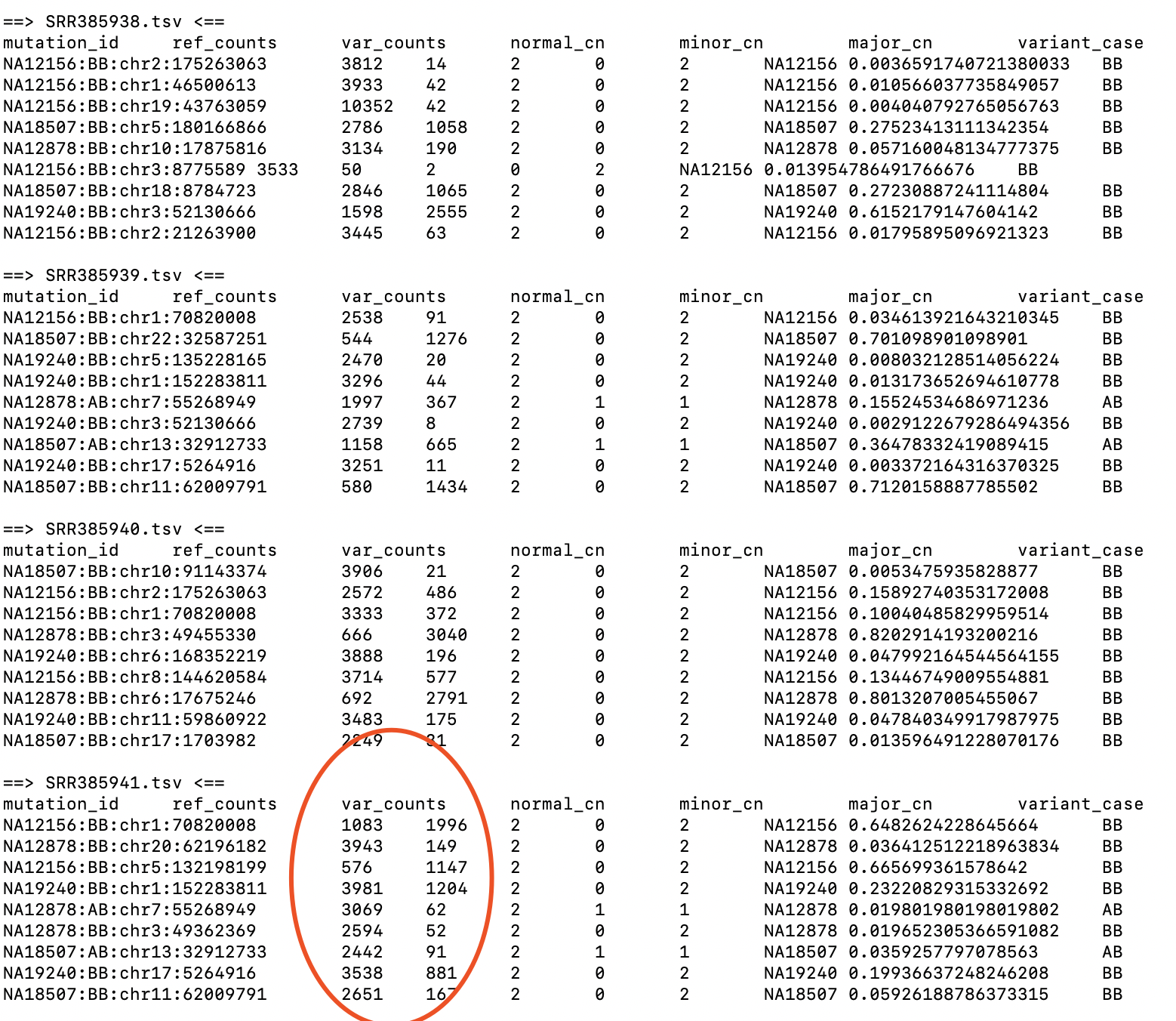
输入的每个样本的tsv文件的格式
tab分隔的6列,需要存在header的文件,包括以下6列
- mutation_id,一个能够识别突变的单一ID,比如chr22:12345或者TP53_chr17:753342
- ref_counts,突变位点的reference reads数
- var_counts,突变位点的variant reads数
- normal_cn,正常population的细胞拷贝数,对于人类常染色体来说是2,对于人类性染色体来说是1或2
- minor_cn, 肿瘤细胞的minor拷贝数,一般从WGSS或者芯片的数据预测出
- major_cn,肿瘤细胞的major拷贝数,一般从WGSS或者芯片的数据预测出
如果你没有minor copy number 和 major copy number,那么minor copy number设为0 (因为很多软件不考虑基因型,所以不给出不同染色体的拷贝情况), 而major copy number设置为预测的总的拷贝数。 除了上述的列,其它列会自动忽略 使用PyClone run_analysis_pipeline -h查看帮助。
而且值得注意的是,测序深度通常要求很高,比如软件自带的例子里面。
发表软件的文章上面提到需要1000X以上,要求有点高哦!
一般来说,运行速度还行,十分钟左右吧,结果如下:
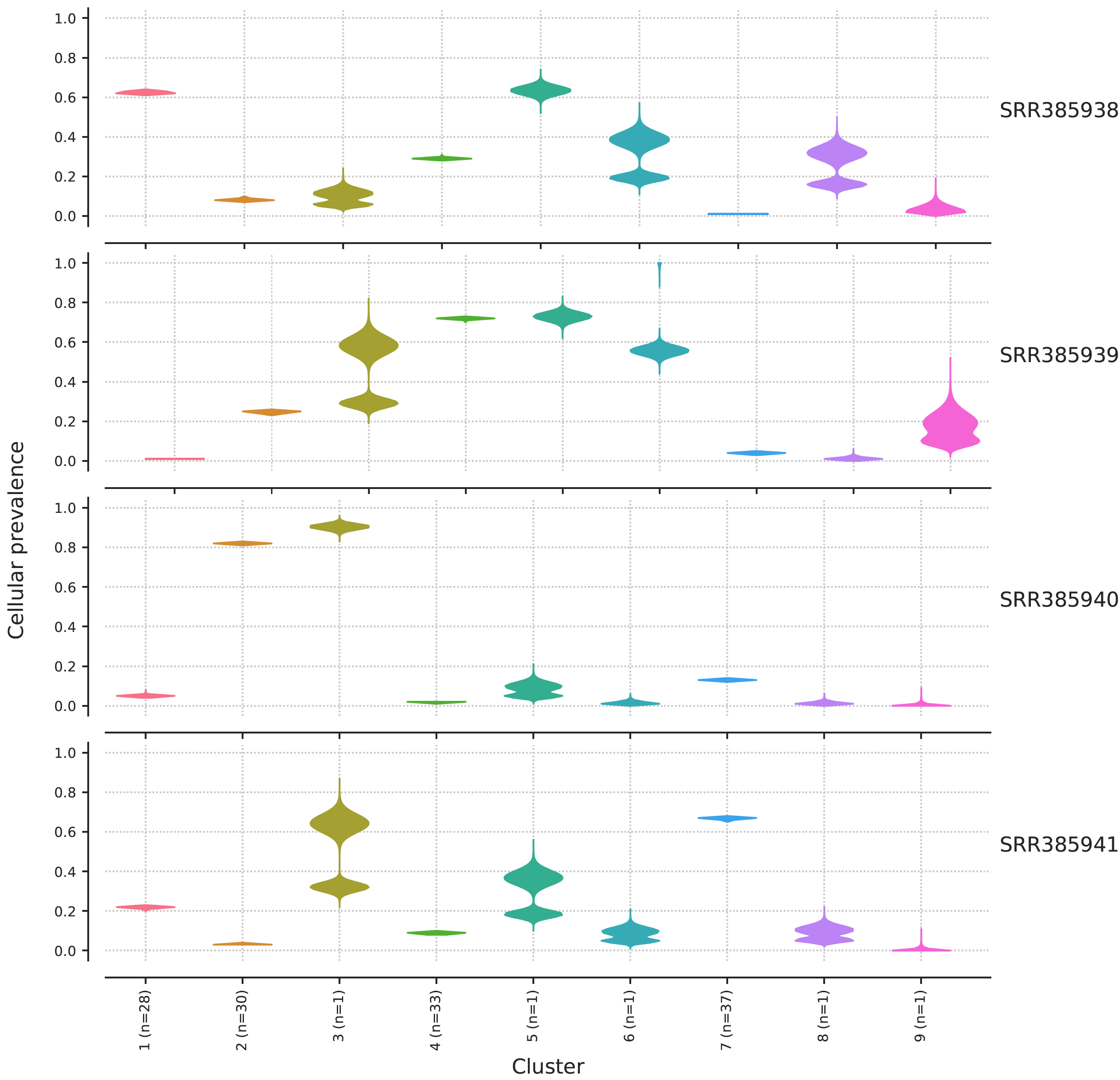
加入这个病人有4个测序文件,就可以看到这个病人总共是有9个亚克隆,然后这个病人的4个不同部位或者不同时间段的主克隆都不一样,比例也很清晰的给出来了。
但是呢,这并不是一个真实的病人的数据,也不是一个病人的4个数据,而是模拟数据。
TCGA结果转为pyclone的input
tcga数据库通常可以下载各个癌症的拷贝数变异文件和somatic的maf格式的突变数据。
所以需要首先从maf格式的突变数据文件里面提取前面的3列,即mutation_id,ref_counts,var_counts,然后把normal_cn全部设置为2,而minor_cn全部设置为0,最后一列真正的CNV值,直接使用bedtools等工具把突变的坐标映射到拷贝数变异文件即可。
这个坐标映射的思想,我在也讲解过。
因为是每个样本独立分析,如果是在R里面代码可以是:
o = findOverlaps(som_gr,this_cnv)
t1=som_gr[queryHits(o)]
t2=this_cnv[subjectHits(o)]
t1=as.data.frame(t1)
t1$cnv=mcols(t2)[,2]
## 想办法制作 6 列的 内容输出。
colnames(out)=c('mutation_id' ,'ref_counts' ,'var_counts' ,
'normal_cn', 'minor_cn', 'major_cn')
out$major_cn=round(2^(out$major_cn+1))
write.table(out,'for_pyclone.tsv',sep = '\t',row.names = F,quote = F)
上面的代码有点难度,建议花费10个小时理解,这样的文件就可以输出,给pyclone作为input咯。
运行pyclone到一个TCGA样本
有了上面的文件,运行pyclone倒是很容易,但是呢,这样意义其实不大,因为测序深度都不够高,同一个病人虽然也可以跑软件得到多个亚克隆。
比如一篇文章 Oncotarget. 2016 Mar , 题目是:Pan-cancer analysis of intratumor heterogeneity as a prognostic determinant of survival ,研究纳入TCGA数据库的9个癌症种类,3300个患者,就是对每个数据走pyclone流程, 拿到压克隆数量来代表异质性。
鱼图
pyclone的结果可以无缝连接到:https://bioconductor.org/packages/release/bioc/vignettes/timescape/inst/doc/timescape_vignette.html
参考:
- pyclone usage:https://bitbucket.org/aroth85/pyclone/wiki/Usage
- pyclone文献:https://www.nature.com/articles/nmeth.2883
- suprahex处理pyclone结果:http://suprahex.r-forge.r-project.org/demo-PyClone.html