做GSEA分析你的基因到底该如何排序
大家都知道,GSEA最重要的就是数据集的所有背景基因按照某种指标排序好,这样才能说明你感兴趣的基因集是否在背景基因集里面出现了统计学显著的富集情况,如下:
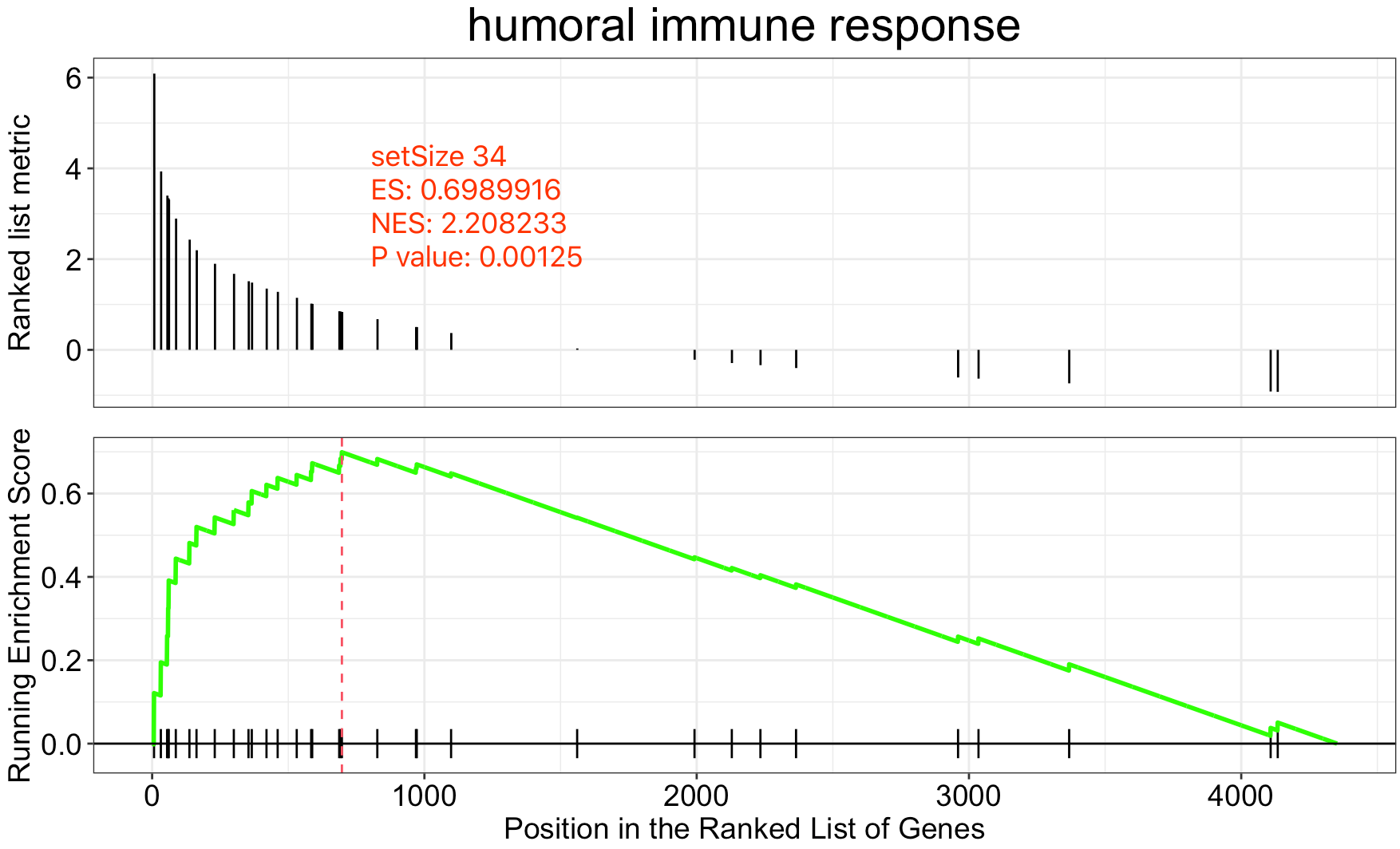
很明显,我们感兴趣的这个基因集,也就是 humoral immune response 有34个基因,而且是显著性的分布在我们的4000多个背景基因的头部,经过统计学公式计算得到了它的一些指标,说明了其显著性。
但是这里面隐藏着一个很有趣的细节,就是背景基因到底是应该如何排序呢? 而java版的GSEA软件提供6种排序方法,包括:
- signal-to-noise ratio (S2N)
- ratio of average expression from two classes (Ratio)
- T-test statistic (T-test)
- the Pearson correlation coefficient for quantitative studies
毕竟,有一个哲人曾经说过:所有的统计学模型都是错的,但总有一些是好用的,所以探究哪一个更好用是我们永恒不变的追求。
12种排序方法
在文章:2017 https://doi.org/10.1186/s12859-017-1674-0 里,作者分析了 28个数据集,使用了 16 种排序方法,做了非常详细的统计学指标比较分析。最后的结论是:
The absolute value of Moderated Welch Test has the best overall sensitivity and Minimum Significant Difference has the best overall specificity of gene set analysis.
感兴趣的朋友可以努力翻译弄懂这篇文章,非常值得初学者耗费心力仔细研读,如果你愿意深究它,请务必邮件联系我,让我知道,我希望认识一些优秀的小伙伴。
这16种排序方法如下表:
| T-test | ||
|---|---|---|
| MWT | ||
| \ | MWT\ | |
| MSD | ||
| S2N | ||
| \ | S2N\ | |
| WAD | ||
| \ | WAD\ | |
| Difference | ||
| Ratio | ||
| log2(Ratio) | ||
| FCROS | ||
| SoR | ||
| BWS | ||
| ReliefF | ||
| ReliefF ranked |
如果是DESeq2的差异分析结果
有人推荐如下指标进行排序:
x$fcsign <- sign(x$log2.fold_change)
x$logP=-log10(x$p_value)
x$metric= x$logP/x$fcsign
当然这是一个参考而已。