一些单细胞转录组R包的对象
ExpressionSet
Bioconductor的ExpressionSet是基石,多次讲解过,GEO数据库在R里面下载的就是这个对象。
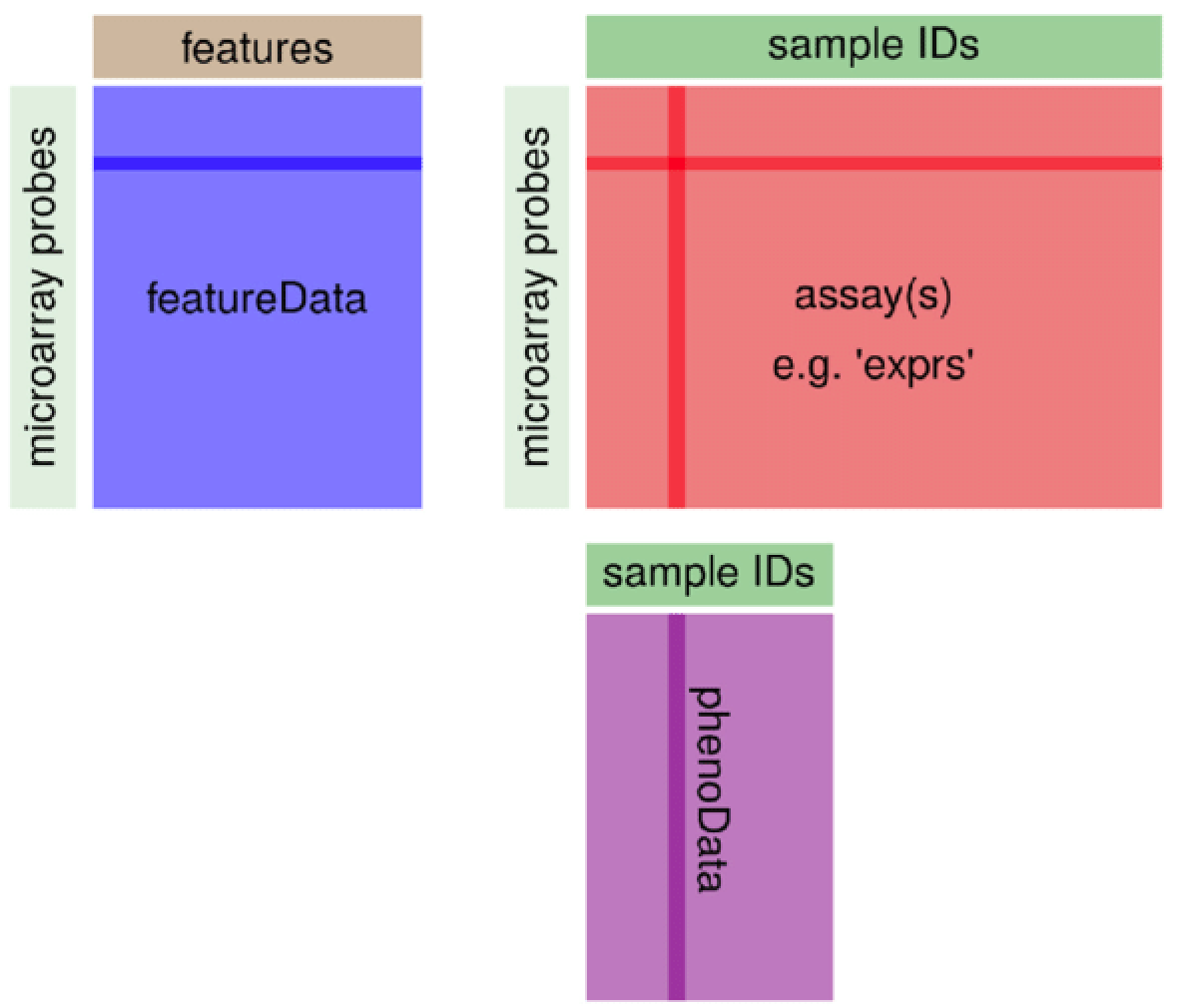
通常不需要自己从头创建。
而单细胞转录组本质上也是转录组,即表达量矩阵的分析,所以后面的R包的对象,其实或多或少借鉴了这个Bioconductor的ExpressionSet对象。
CellDataSet
来自于monocle这个R包,使用其提供的 newCellDataSet() 函数即可创建,创建后的对象组成成分如下
- 表达矩阵:rows as features (usually genes) and columns as cells
- 使用 featureData and phenoData 函数可以获取基因和样本信息
- 其中 expressionFamily指定表达矩阵的归一化形式
归一化形式通常是3种
- tobit() FPKM, TPM
- negbinomial.size() UMIs, Transcript counts from experiments with spike-ins or relative2abs(), raw read counts
- gaussianff() log-transformed FPKM/TPMs, Ct values from single-cell qPCR
先介绍一下monocle需要的用来构建 CellDataSet 对象的三个数据集
- 表达量矩阵
exprs:数值矩阵 行名是基因, 列名是细胞编号. - 细胞的表型信息
phenoData: 第一列是细胞编号,其他列是细胞的相关信息 - 基因注释
featureData: 第一列是基因编号, 其他列是基因对应的信息
并且这三个数据集要满足如下要求:
表达量矩阵必须:
- 保证它的列数等于
phenoData的行数 - 保证它的行数等于
featureData的行数
而且
phenoData的行名需要和表达矩阵的列名匹配featureData和表达矩阵的行名要匹配featureData至少要有一列”gene_short_name”, 就是基因的symbol
准备Monocle对象需要的phenotype data 和 feature data 以及表达矩阵,从 scRNAseq 这个R包里面提取这三种数据。
library(scRNAseq)
## ----- Load Example Data -----
data(fluidigm)
# Set assay to RSEM estimated counts
assay(fluidigm) <- assays(fluidigm)$rsem_counts
ct <- floor(assays(fluidigm)$rsem_counts)
ct[1:4,1:4]
sample_ann <- as.data.frame(colData(fluidigm))
gene_ann <- data.frame(
gene_short_name = row.names(ct),
row.names = row.names(ct)
)
pd <- new("AnnotatedDataFrame",
data=sample_ann)
fd <- new("AnnotatedDataFrame",
data=gene_ann)
sc_cds <- newCellDataSet(
ct,
phenoData = pd,
featureData =fd,
expressionFamily = negbinomial.size(),
lowerDetectionLimit=1)
sc_cds
这样就完成了CellDataSet对象的从头构建,它是后续分析的基石。
SingleCellExperiment
同样的也是那些信息:Different quantifications (e.g., counts, CPMs, log-expression) can be stored simultaneously in the assays slot. Row and column metadata can be attached using rowData and colData, respectively.
主要是scater包采用,也是可以从头构建。
创建代码如下:
library(scRNAseq)
## ----- Load Example Data -----
data(fluidigm)
# Set assay to RSEM estimated counts
assay(fluidigm) <- assays(fluidigm)$rsem_counts
ct <- floor(assays(fluidigm)$rsem_counts)
ct[1:4,1:4]
table(rowSums(ct)==0)
# 这里使用原始表达矩阵,所以有很多基因在所有细胞均无表达量,即表现为没有被检测到,这样的基因是需要过滤掉的。
pheno_data <- as.data.frame(colData(fluidigm))
## 这里需要把Pollen的表达矩阵做成我们的 scater 要求的对象
#data("sc_example_counts")
#data("sc_example_cell_info")
# 你也可以尝试该R包自带的数据集。
# 参考 https://bioconductor.org/packages/release/bioc/vignettes/scater/inst/doc/vignette-intro.R
sce <- SingleCellExperiment(
assays = list(counts = ct),
colData = pheno_data
)
sce
#
#
后面所有的分析都是基于 sce 这个变量 ,是一个 SingleCellExperiment 对象,被很多单细胞R包采用。
seurat 对象
主要是seurat包采用该对象,个人觉得并不是很方便,并不是上面的SingleCellExperiment 对象。
library(scRNAseq)
## ----- Load Example Data -----
data(fluidigm)
# Set assay to RSEM estimated counts
assay(fluidigm) <- assays(fluidigm)$rsem_counts
ct <- floor(assays(fluidigm)$rsem_counts)
ct[1:4,1:4]
names(metadata(fluidigm))
meta <- as.data.frame(colData(fluidigm))
counts <- ct
identical(rownames(meta),colnames(counts))
Pollen <- CreateSeuratObject(raw.data = counts,
meta.data =meta,
min.cells = 3,
min.genes = 200,
project = "Pollen")
Pollen
并不一定要对象
有些单细胞转录组R包,就没有封装为特殊的对象,而是简单的list即可,比如M3Drop这个单细胞转录组R包:
library(scRNAseq)
## ----- Load Example Data -----
data(fluidigm)
ct <- floor(assays(fluidigm)$rsem_counts)
ct[1:4,1:4]
sample_ann <- as.data.frame(colData(fluidigm))
counts=ct
library(M3Drop)
Normalized_data <- M3DropCleanData(counts,
labels = sample_ann$Biological_Condition ,
is.counts=TRUE, min_detected_genes=2000)
dim(Normalized_data$data)
length(Normalized_data$labels)
class(Normalized_data)
str(Normalized_data)
那你需要对象吗
你觉得对象重要吗,需要嘛?