为了分析不同类型、组织起源肿瘤的共性、差异以及新课题。TCGA于2012年10月26日-27日在圣克鲁兹,加州举行的会议中发起了泛癌计划。参考:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6000284/ 为此我也录制了系列视频教程在:TCGA知识图谱视频教程(B站和YouTube直达)
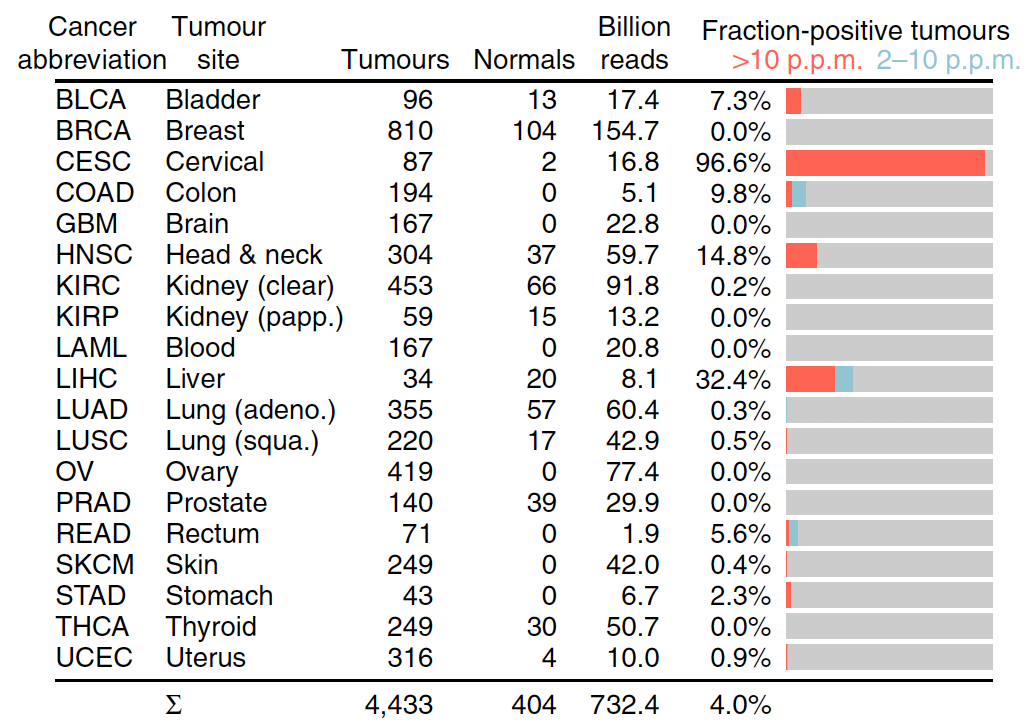
本文发表的杂志还算中规中矩啦,在 Nat Commun. 2013; 题目是:The landscape of viral expression and host gene fusion and adaptation in human cancer. 可能是因为关注的是病毒,所以引用不多,不到200次。基于的mRNA_seq测序数据,系统性的研究了4,433 tumours and 19 cancer types,发现了不少病毒整合位点及基因。
文献解读属于100篇泛癌研究文献系列,首发于:http://www.bio-info-trainee.com/4132.html
样本量如下
这次涉及的样本量和癌症种类多一点,是 4,433 tumours and 19 cancer types, 主要基于的mRNA_seq测序数据,而且不是基于表达矩阵,所以需要跟TCGA团队申请原始数据。
病毒整合分析流程
主要是从mRNA_seq测序数据里面提取那些无法比对到人类参考基因组的序列
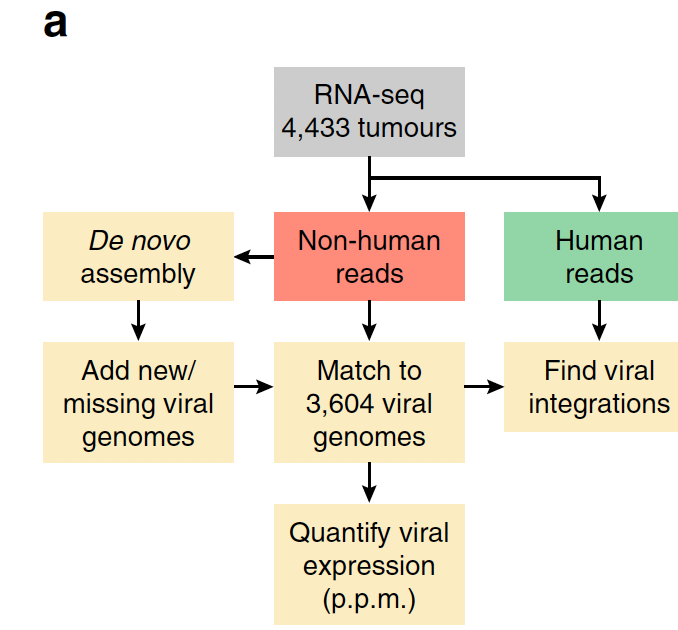
病毒参考基因组
3,065 non-human viral genomes, 下载于 RefSeq collection of viral genomes (n.3,590 excluding bacterial phages), downloaded on 19 Dec 2012.
使用的是bowtie软件,参数是 up to 2 mismatches and a maximum of 25 alignments
其中HNSC的HPV情况是TCGA团队实验确定的
关于病毒基因组组装
使用了 SOAPdenovo 软件,得到的contigs再使用BLAST
病毒基因组整合
这样的整合会影响拷贝数变异和表达量变化,所以可以分组后找差异基因:

使用阈值:a false discovery rate (q) < 0.05 and with an absolute log2 median expression ratio > 2. 可以拿到 Five hundred and ninety-seven host genes
而且病毒阳性样本的表达相关性可以跨越癌症种类聚在一起:
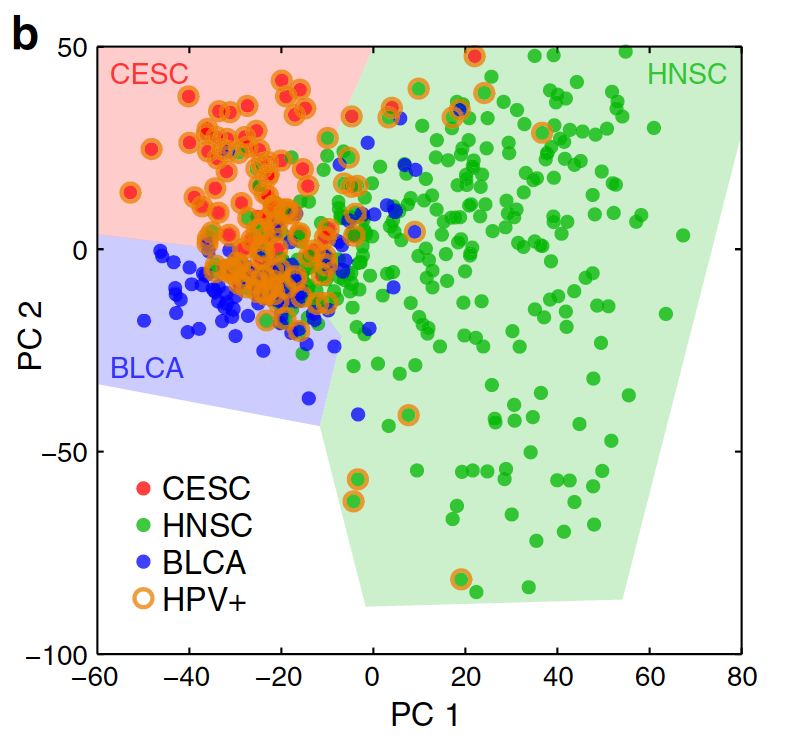
PCA analysis of tumour mRNA expression profiles in CESC, HNSC and BLCA. Although there were systematic expression differences between cancer types, HPV-positive tumours clustered together regardlessof type.
后记
本研究的工作量还蛮大的,毕竟下载了TCGA数据库的RNA-seq的原始数据,然后自主数据分析流程,绝大部分实验室是做不到这一点的。
本文献解读属于100篇泛癌研究文献系列,首发于:http://www.bio-info-trainee.com/4132.html