为了分析不同类型、组织起源肿瘤的共性、差异以及新课题。TCGA于2012年10月26日-27日在圣克鲁兹,加州举行的会议中发起了泛癌计划。参考:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6000284/ 为此我也录制了系列视频教程在:TCGA知识图谱视频教程(B站和YouTube直达)
发表于普通杂志:Mol Cancer Res. 2019 Feb; 文章是:Molecular Correlates of Metastasis by Systematic Pan-Cancer Analysis Across The Cancer Genome Atlas. 系统性的研究了TCGA数据库的11种癌症的 4,473 primary tumor samples and 395 tumor metastasis samples ,发现不同癌症的 转移和原位癌的表达差异都很大,不同癌症有一些overlap情况,当然除了比较mRNA-seq数据,还有miRNAs,RPPA, DNA methylation 的数据的比较探索。还利用了 Gene expression data (TPM values) from GTEx Analysis version 7 数据库,也有一些GEO数据库的,比如GSE110590。
文献解读属于100篇泛癌研究文献系列,首发于:http://www.bio-info-trainee.com/4132.html
差异表达
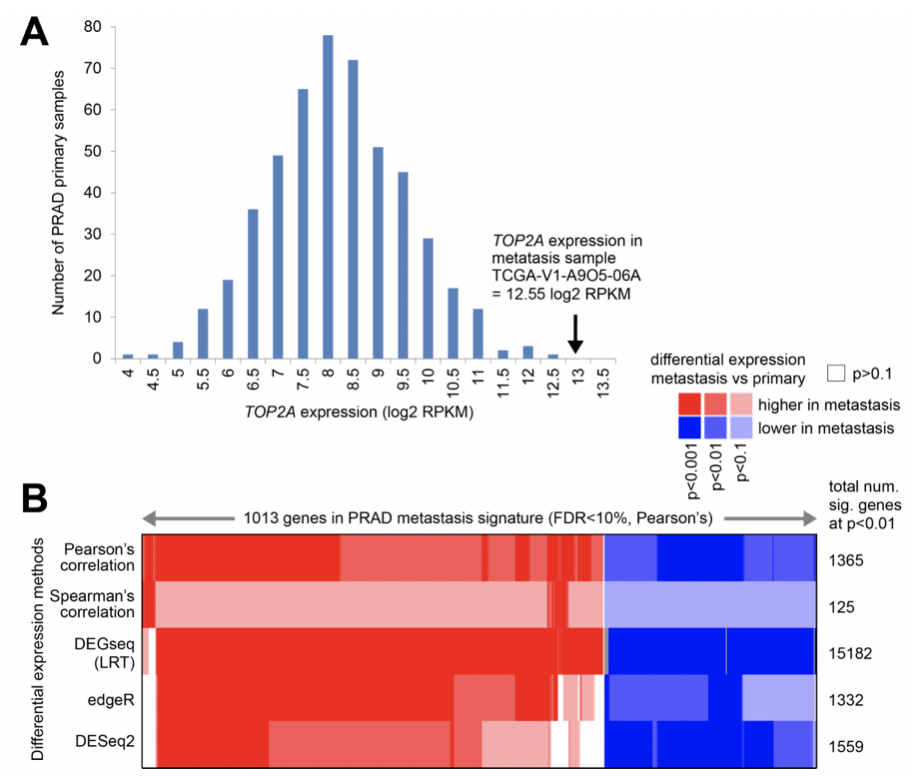
样本量如此悬殊,作者居然也做了差异分析
作者采用了多种统计学算法来寻找差异基因:
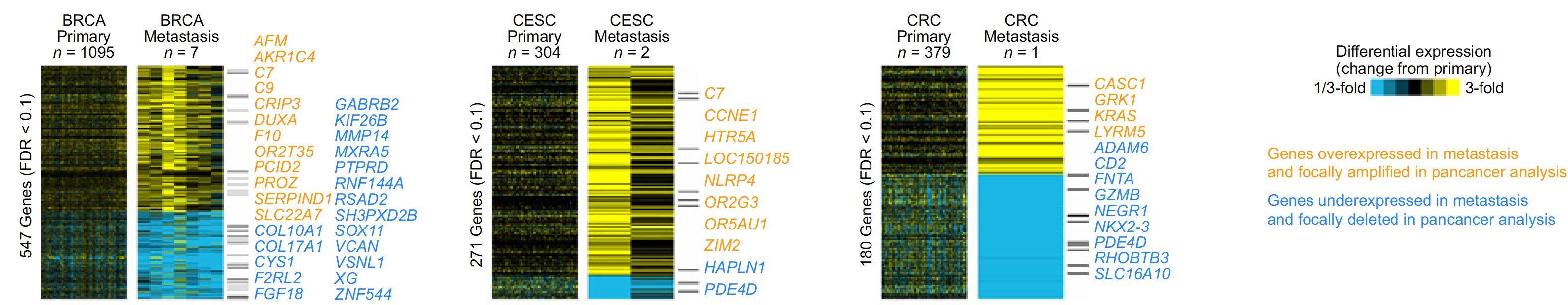
不同癌症的上下调基因的overlap情况如下:
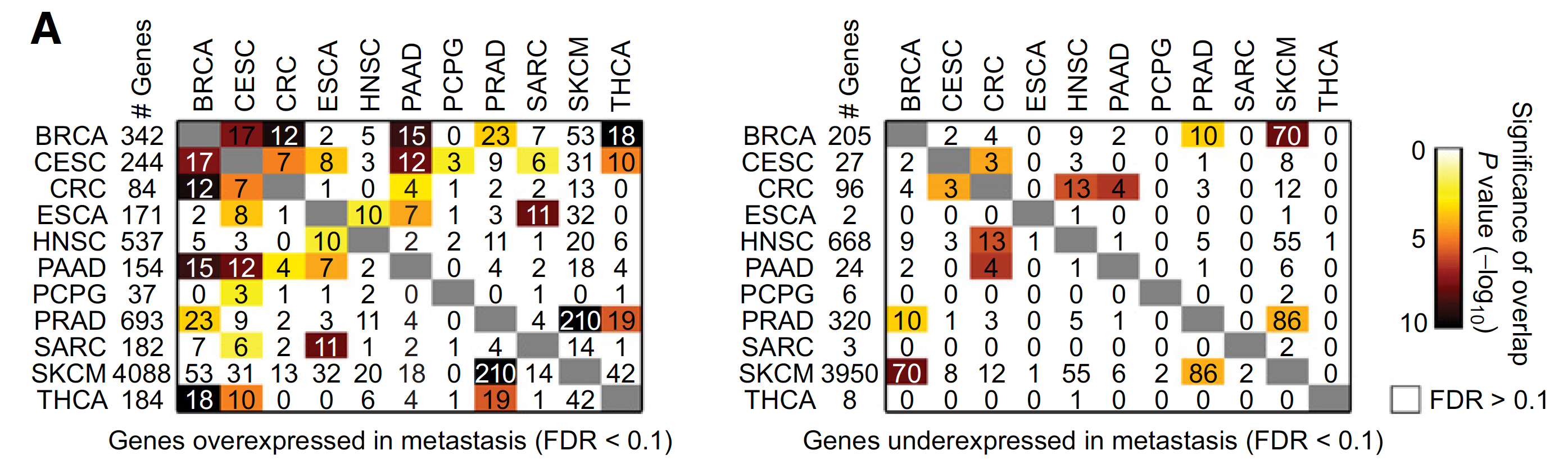
不同癌症的上下调基因集的overlap情况:
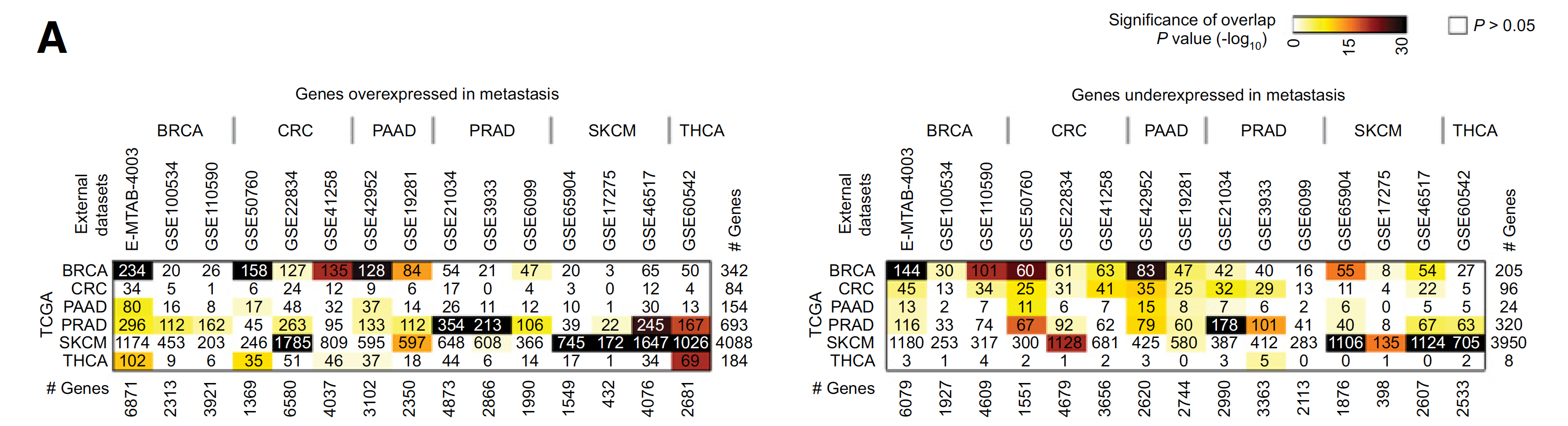
TCGA数据库和GEO数据库的比较
如下:
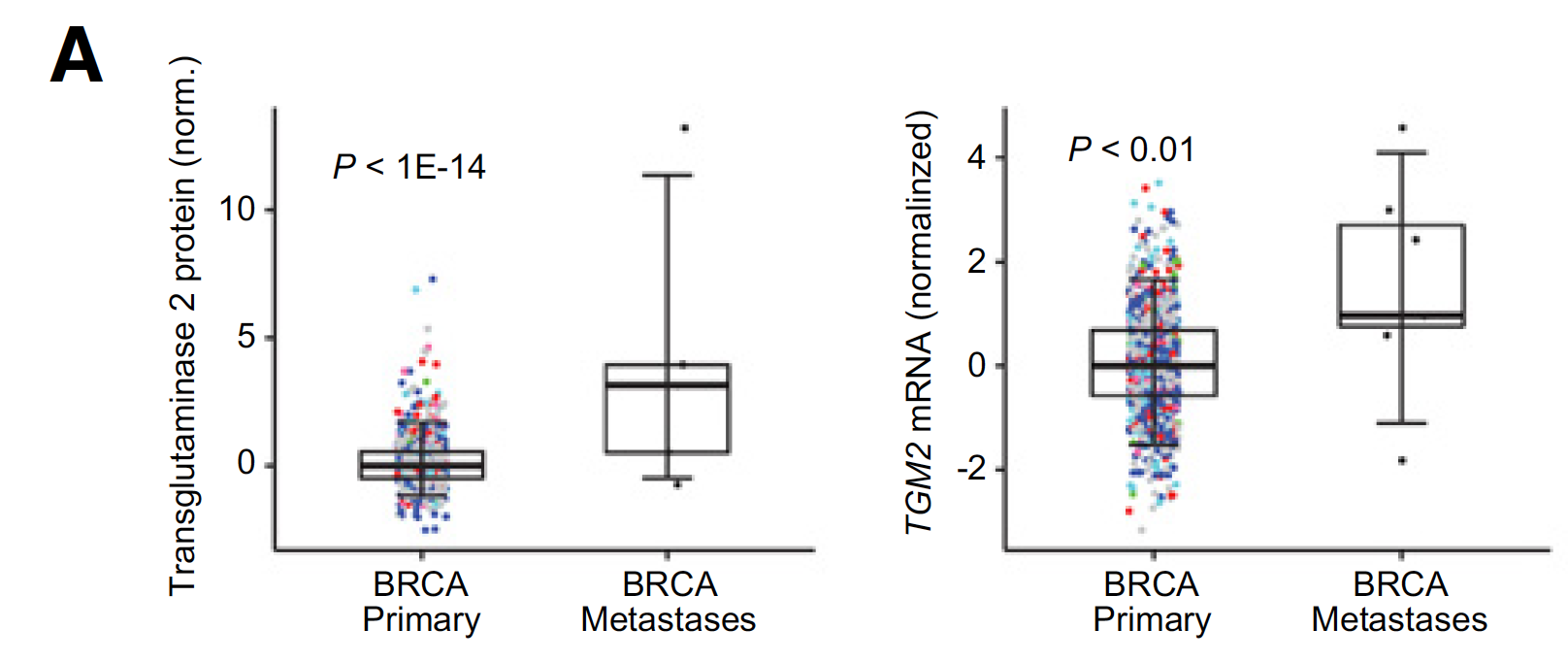
蛋白质芯片数据的泛癌比较
RPPA proteomic data involved 218 features and four cancer types (BRCA, PCPG, SKCM, and THCA) with metastasis profiles.
下面是其中一个例子,蛋白和编码其的基因都是显著差异
miRNA表达数据的泛癌比较
For each cancer type examined, the correlations with metastasis for RPPA (Reverse Phase Protein Array) and microRNA features represented in TCGA. Also included are mRNA:microRNA pairings, as defined by both a previously identified miRNA-target interaction (as cataloged by miRTarBase Release 7.0) and significant differential expression in metastasis (FDR<0.1) for both mRNA and microRNA, in opposite directions from each other (mRNA up:microRNA down or mRNA down:microRNA up).
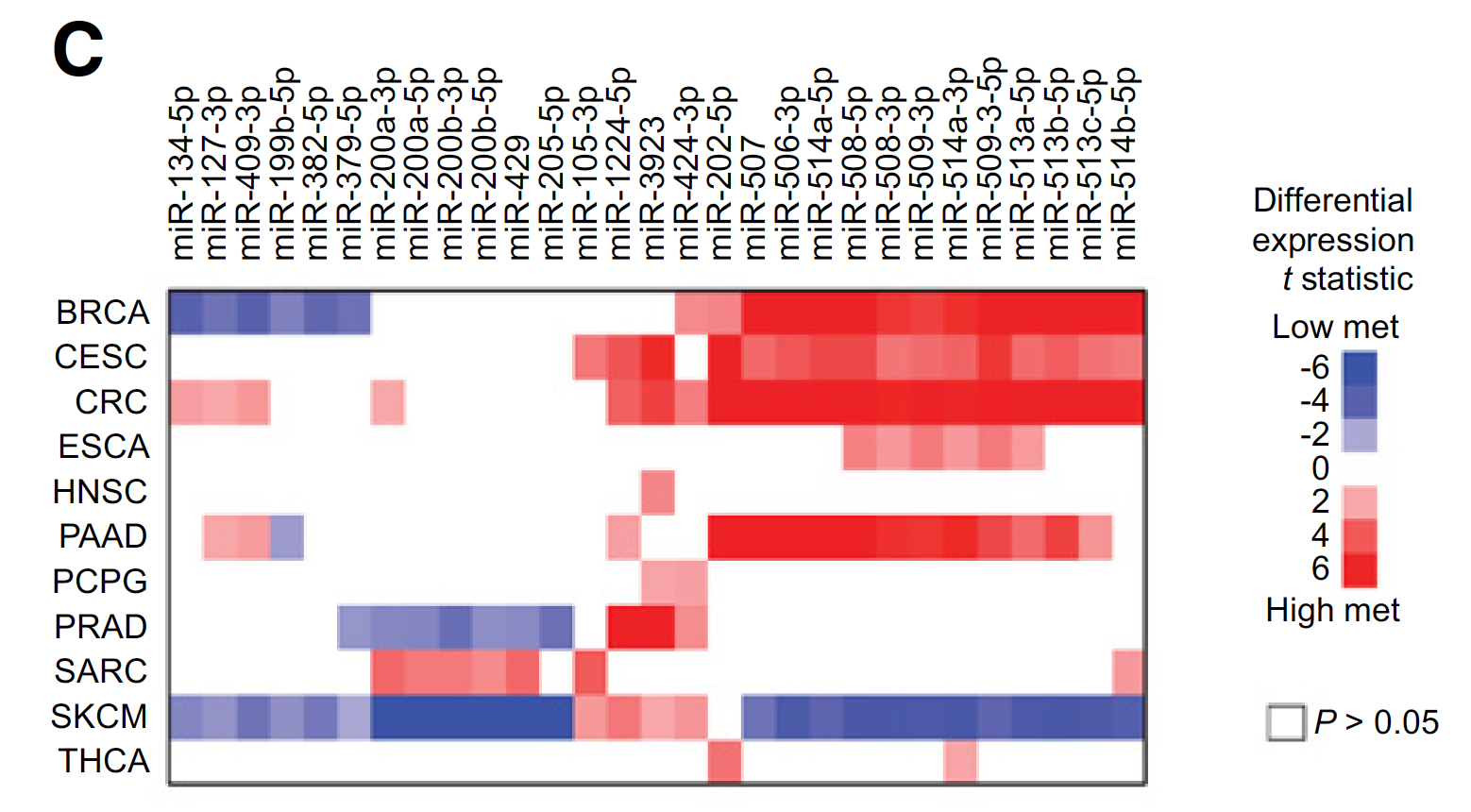
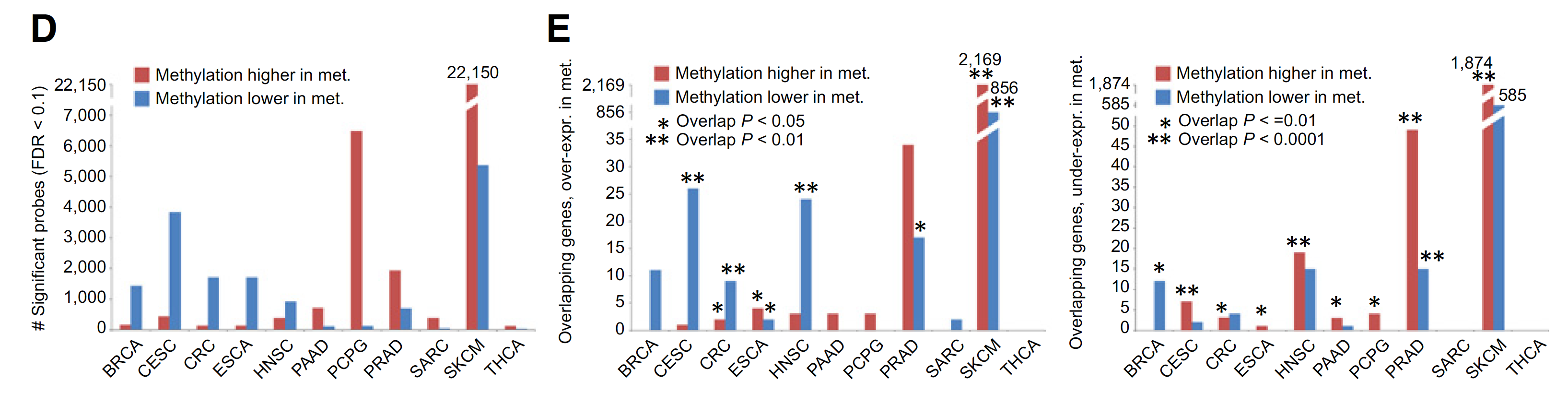
DNA甲基化芯片数据的泛癌比较
For each cancer type, top metastasis-associated DNA methylation CpG Island features, selected using Pearson’s correlation (logit-transformed values) with Storey and Tibshirini estimate of False Discovery Rate (FDR) of <10%. Differential mRNA statistics (metastasis versus primary) corresponding to the associated genes are also included.
主要关注:CpG Islands (by Illumina 450K array, 150K CpG Island probes)
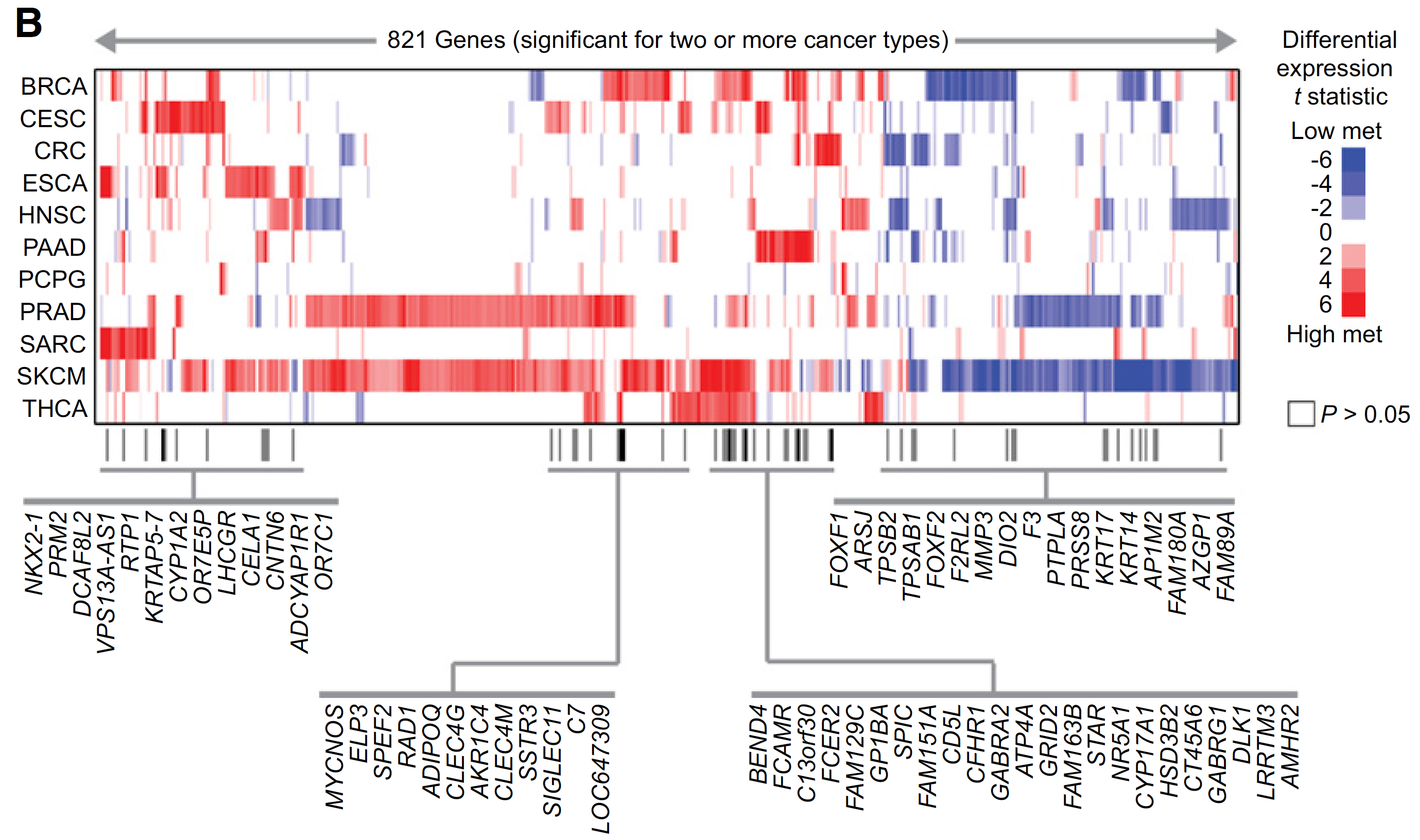
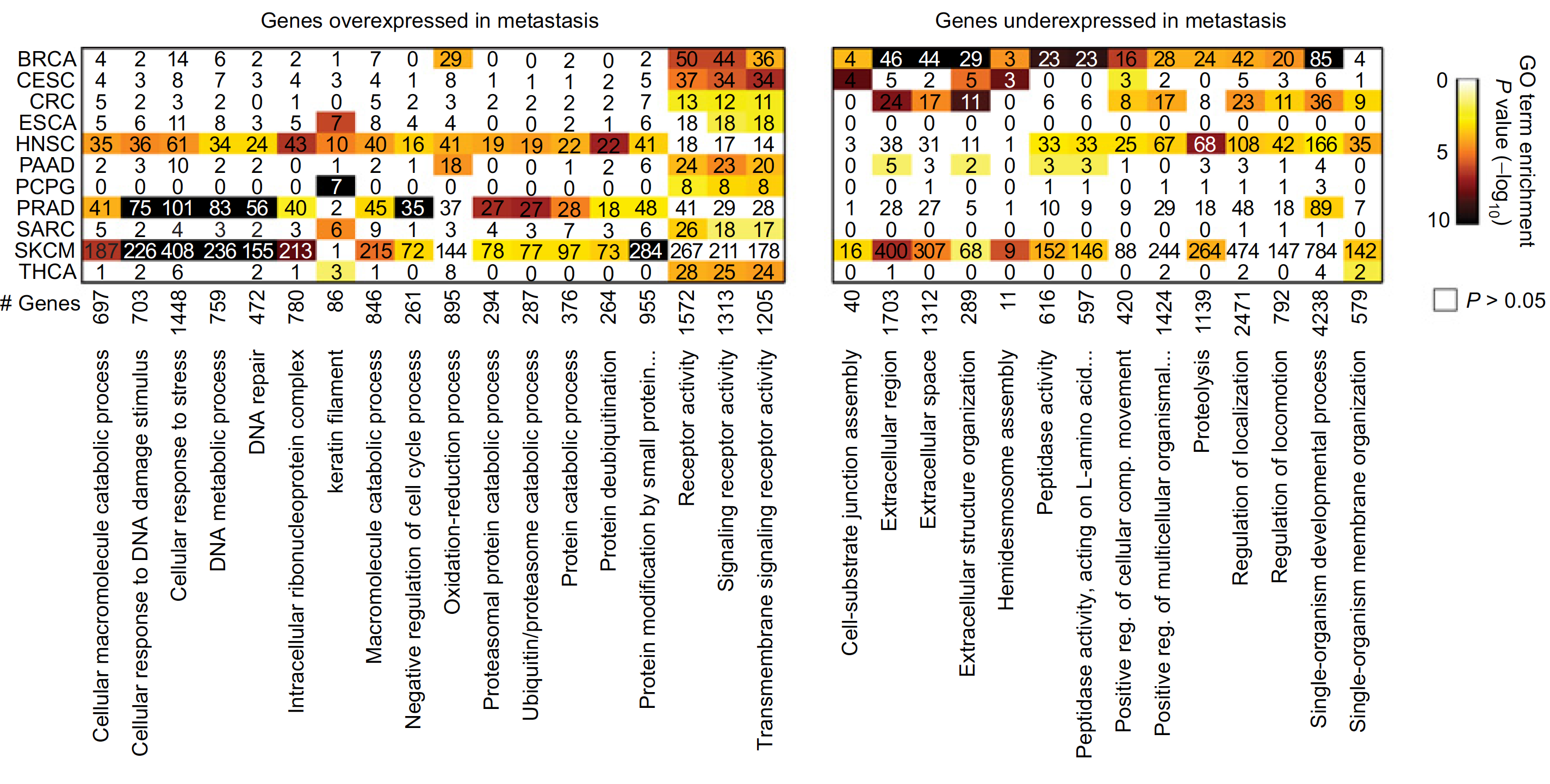
图展示差异甲基化位点和差异表达基因的overlap情况,如下;
定下 metastasis signature
这里并没有使用 miRNAs,RPPA, DNA methylation 的数据,就是纯粹的mRNA-seq数据来获得的 metastasis signature
A set of 821 genes were found significant (FDR < 10%) with same direction of change for two or more cancer types
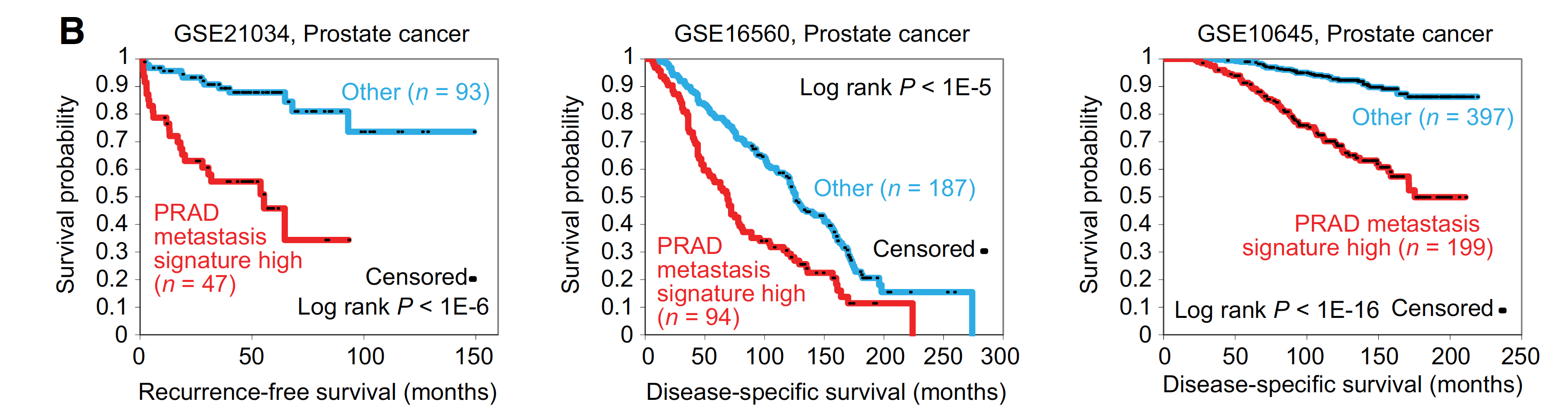
生存分析说明临床意义
比较奇怪的是,这里并没有展示作者自己的821个基因的metastasis signature 在TCGA的生存分析效果,反而是用前列腺癌的GEO数据。
The TCGA-derived prostate cancer metastasis signature in particular could define a subset of aggressive primary prostate cancer.
补充材料
- Supplementary Information - Supplementary Figures and Description of Data Files
- Table S1 - TCGA cancer cases and molecular profiles examined in this study.
- Table S2 - For all genes represented in TCGA RNA-seq datasets, the mRNA-level correlations with metastasis for each cancer type.
- Table S3 - For each cancer type, top metastasis-associated mRNA features, selected using Pearson’s correlation on log-transformed data with Storey and Tibshirini estimate of False Discovery Rate (FDR) of <10%.
- Table S4 - Gene Ontology (GO) term associations for the top metastasis-associated genes for each cancer type.
- Table S5 - For each cancer type examined, the correlations with metastasis for RPPA (Reverse Phase Protein Array) and microRNA features represented in TCGA.
- Table S6 - For each cancer type, top metastasis-associated DNA methylation CpG Island features, selected using Pearson’s correlation (logit-transformed values) with Storey and Tibshirini estimate of False Discovery Rate (FDR) of <10%. Differential mRNA statistics (metastasis versus primary) corresponding to the associated genes are also included.
后记
从流程图来看,本研究并不复杂,也很容易复现出来, 关键是如何提出还有如何挑选数据集。
本文献解读属于100篇泛癌研究文献系列,首发于:http://www.bio-info-trainee.com/4132.html