2018年2月的一篇PNAS文章:Global changes of H3K27me3 domains and Polycomb group protein distribution in the absence of recruiters Spps or Pho 的作者来我们学校面试助理教授,有幸听他完整讲述他们课题组的工作。
这篇文章只有两个共一的作者,一个负责全部的实验环节,一个负责全部的数据分析环节,所以作者的确是有真才实学的,佩服!
为了探索PcG的招募机制
背景知识:
肯定是需要自行搜索了解一下PRC复合物相关知识咯,而且这个复合物在不同物种稍微有点不一样,是表观领域的热点。
- Polycomb group (PcG) proteins
- Polycomb response elements (PREs)
- Pho
- Cg
- Spps
- third instar larval stage - FlyBase
- | Supplementary file | Size | Download | File type/resource |
| ———————————————— | ———— | —————————————————————————————— | ——————————— |
| GSE102339_BED_FILES.tar.gz | 3.4 Mb | (ftp)(http) | TAR |
| GSE102339_normalized_fpkm.txt.gz | 552.1 Kb | (ftp)(http) | TXT |
结论:PcG recruiters, the PRC2 component E(z), and the PRC1 components Psc and Ph cobind thousands of active genes outside of H3K27me3 domains.文章主要分析点
这篇文章可以说是RNA-seq和ChIP-seq数据分析结合的典范了,我们后期会录制视频课程和详细推文发布在生信技能树公众号。
首先需要参考生信技能树B站的RNA-seq和ChIP-seq数据分析视频教程,对本文数据拿到peaks文件和表达矩阵。
全部教学视频在:https://space.bilibili.com/338686099/#/peaks的基因元件(启动子,外显子,内含子)注释
主要是针对peaks文件进行注释,结果gtf文件即可,有R包可以做。
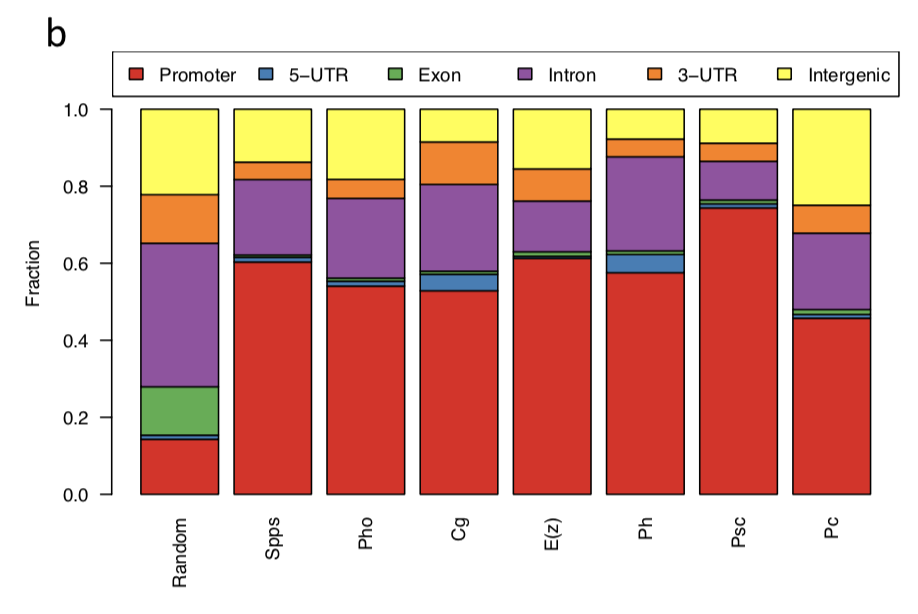
Figure S1. Global characteristics for the binding of PcG proteins and recruiters多个peaks的bed文件找overlap(教程示例)
同样是很简单的R包操作peaks文件,需要注意的是,文章的3个peaks文件,首先统一经过了H3K27me3的过滤,然后再取交集,如下图:
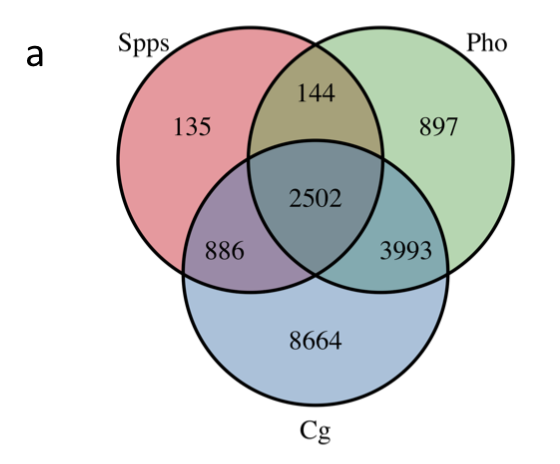
- 介绍什么是chip-seq的peaks
- 介绍bed格式
- 介绍ChIPpeakAnno包
- 介绍findOverlapsOfPeaks函数
- 介绍韦恩图可视化
IGV可视化bam和bigwig文件
这个主要是靠IGV技巧,我在生信技能树的小技巧视频课程详细讲解过,需要调整的细节很多。
bam文件相关性(deeptools)
这是deeptools自带的图片,非常简单。
Figure S6 The correlation of changes of H3K27me3 intensity after knockout of different recruiterschip-seq的信号值矩阵的差异分析
差异分析,这里把peaks的区域当做是基因来,就可以拿到信号值矩阵。
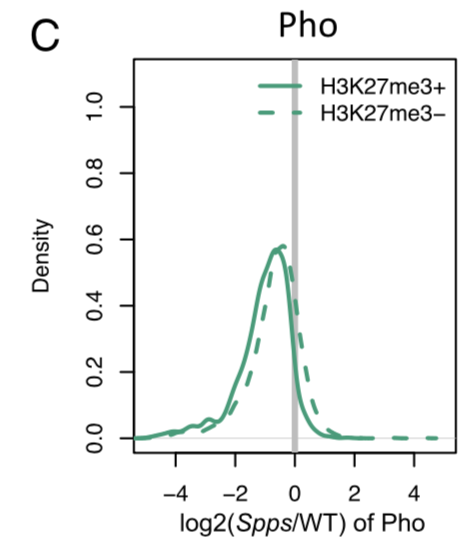
Figure S7 The differential binding of PcG proteins and recruiters after the disruption of SppsRNA-seq的表达矩阵的差异分析,两个差异分析结果的比较
这里只是很简单的把差异分析的logFC进行散点图可视化。
Figure S13 Differential gene expression after the disruption of Pho and Spps are moderately correlatedRNA-seq的bam的IGV可视化看基因敲除效果
这个是非常有必要的图表,说明了实验的可靠性。

Figure S12 Decreased expression of Pho and Spps in the corresponding mutantschip-seq和RNA-seq的IGV结合可视化
首先需要找到值得可视化的区域或者基因来辅助阐述自己的生物学故事。
单个bam测序比对文件按照单个bed坐标文件进行信号值计算及可视化
介绍bam文件,bed文件,信号值,deeptools软件,最后出图
只需要一个bed文件,一个bam文件,使用deeptools即可。
多个bam测序比对文件按照多个bed坐标文件进行信号值计算及可视化
在单个bam文件和单个bed文件出图基础升华即可,把deeptools吃透。
两个bam测序比对文件的信号值比例
数据处理复现
数据分析需要下载参考基因组;https://asia.ensembl.org/Drosophila_melanogaster/Info/Index
测序数据都在:GSE102339:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE102339和[SRP114984](https://www.ncbi.nlm.nih.gov/sra?term=SRP114984) 总共是109G数据,所以根据服务器配置自由调整需要练习的数据。
GSM2734944 ChIP-Seq_Ez_WT_1
GSM2734945 ChIP-Seq_Ez_WT_2
GSM2734946 ChIP-Seq_Ez_WT_3
GSM2734991 RNA-Seq_WT_1
GSM2734992 RNA-Seq_WT_2
GSM2734993 RNA-Seq_WT_3
根据作者给出的ID号,使用prefetch下载,中国大陆需要配套IBM的aspera假设。
cat SRR_Acc_List.txt | while read id;do (nohup ~/biosoft/sratoolkit/sratoolkit.2.9.2-centos_linux64/bin/prefetch $id -X 100G -O sra/ & );done
下载得到的sra文件后走生信技能树B站的RNA-seq和ChIP-seq数据分析视频教程即可。
全部教学视频在:https://space.bilibili.com/338686099/#/