本次分析的文章发表在Plant Cell. 2019 May; 题为‘High-temporal-resolution Transcriptome Landscape of Early Maize Seed Development’的学术论文。该研究通过对玉米珠心(包括胚囊)进行RNA-seq分析,绘制了高时间分辨率的玉米籽粒发育早期转录组图谱。 需要注意的是 days after pollination (DAP) 和 hours after pollination (HAP)
早在2008年,就有研究者使用表达芯片技术,探索过6个时间点的玉米种子发育过程的转录调控变化细节。2013和2014也分别有研究团队使用RNA-seq技术探索。具体不是我的研究领域,懒得翻译了。总之,由于之前的转录组研究没有足够的时间分辨率,可能有多个只在特定发育阶段短时间表达、但对籽粒早期发育很重要的基因没有得到解析。作者这个研究是目前为止涉及到的时间点最多的,可以检测到22,790 genes, including 1415 transcription factors (TFs),的动态变化,其中1093 genes, including 110 TFs是有统计学特异性的表达在某个发育阶段。
4个发育阶段
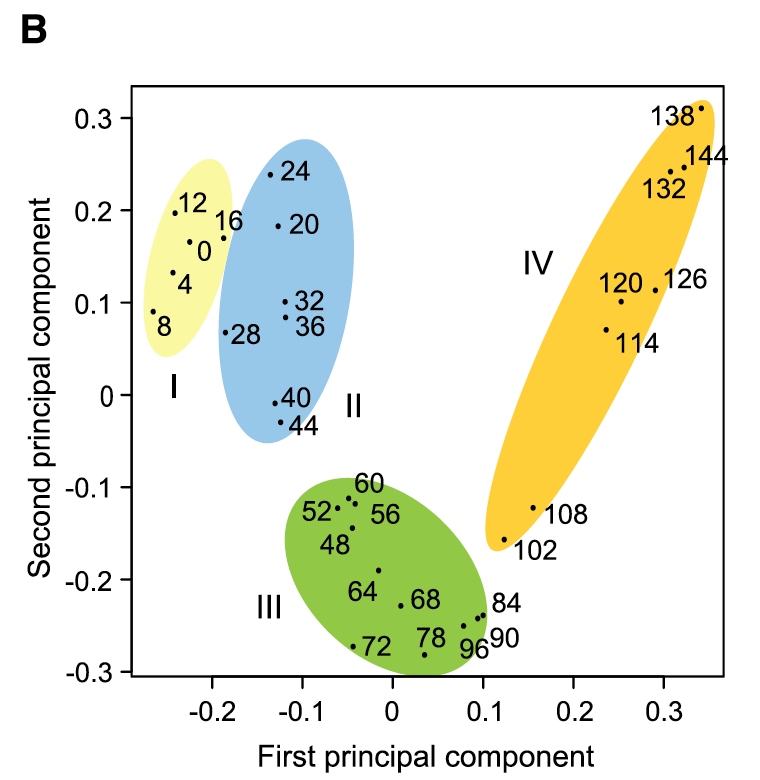
对RNA-seq数据的转录表达谱进行主成分分析还有层次聚类,都可以清晰的看到玉米种子发育早期的31个时间点,可以清晰的分成4个发育阶段:
- 双受精(double fertilization)
- 多核形成(coenocyte formation)
- 细胞化(cellularization)
- 分化(differentiation)
示意图如下:
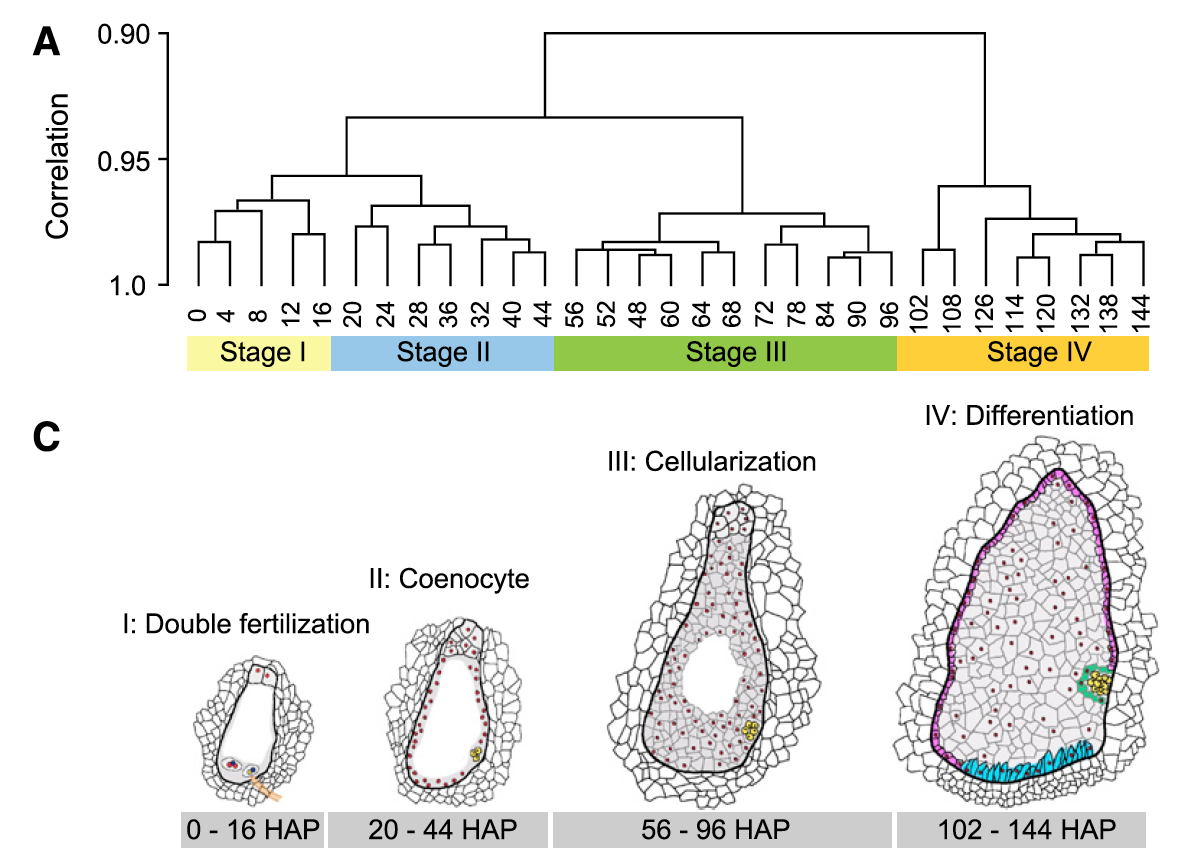
每个阶段都有特异性高表达基因
在玉米种子早期发育阶段共有22790个基因表达,包括1415个转录因子。其中,有1093个基因(包括110个转录因子)只在种子中特异性的表达;这里面大部分基因是在本次研究中被发现的,而且它们具有高度的时间表达特性,也就是说它们只在种子发育的特定阶段表达。后面会进行细致的注释和解释。
可以看到,在授粉后的前16小时、胚乳游离核期、细胞化和分化阶段分别鉴定到了160、22、112和569个籽粒特异性基因。
| Developmental Stage | No. of Genes/TFs | No. of Specific Genes/TFs |
| :———————————————————- | :—————————- | :—————————————— |
| Around double fertilization (0 ∼ 16 HAP) | 4,453/414 | 160/18 |
| Coenocyte (20 ∼ 44 HAP) | 1,285/53 | 22/1 |
| Cellularization (48 ∼ 96 HAP) | 2,569/125 | 112/7 |
| Differentiation (102 ∼ 144 HAP) | 3,614/224 | 569/60 |
| Other | 10,869/599 | 230/24 |
| Total | 22,790/1,415 | 1,093/110 |
以表达量随着时间变化的折线图表示:
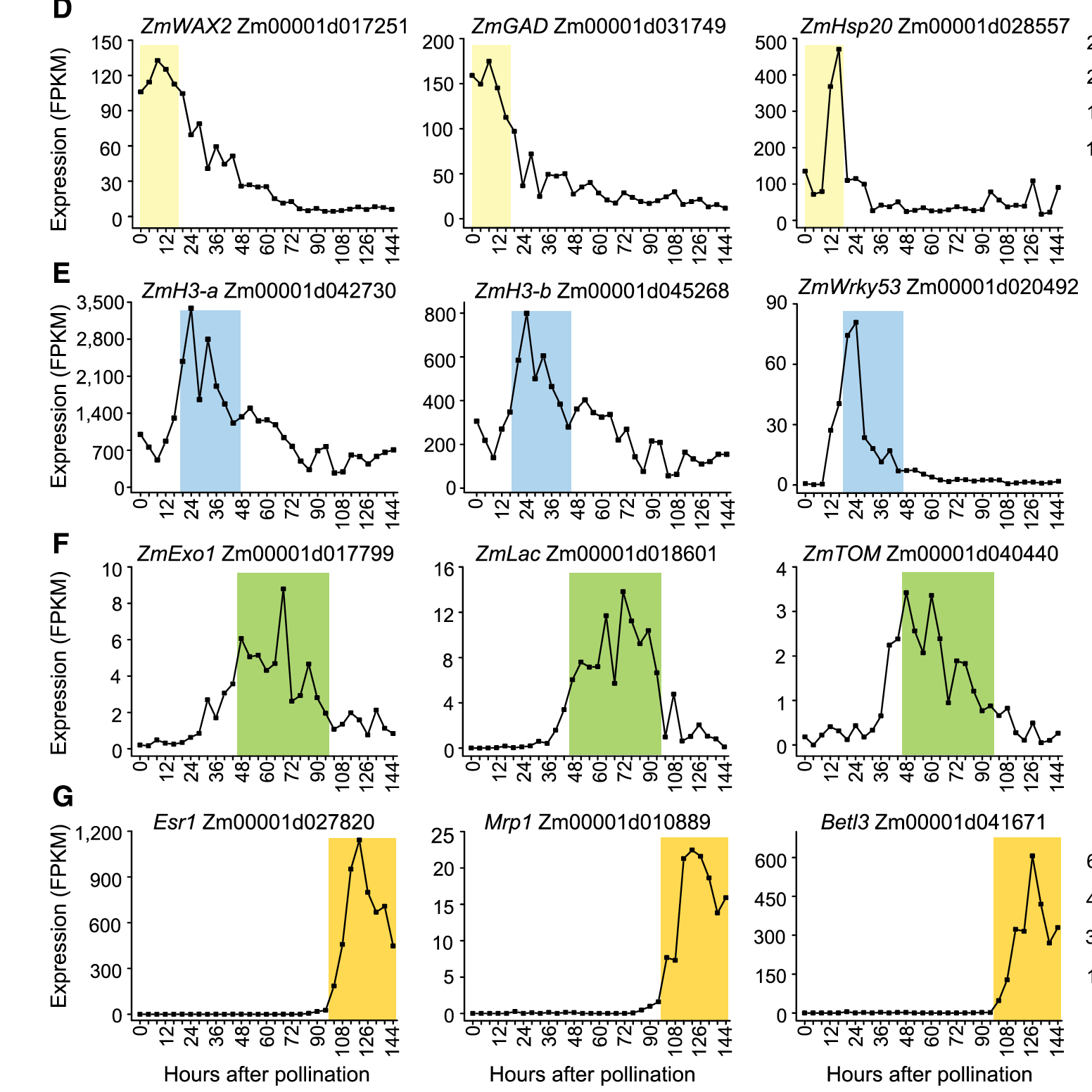
这里,其实可以借鉴单细胞转录组的分析及统计可视化技巧,比如在PCA图上面标记表达量热图,大家可以根据我的提示来尝试一下:
每个发育阶段特异性表达基因的功能注释
可以把每个发育阶段与其它数据进行差异分析,拿到独特的差异基因列表,然后绘图可视化展现如下:
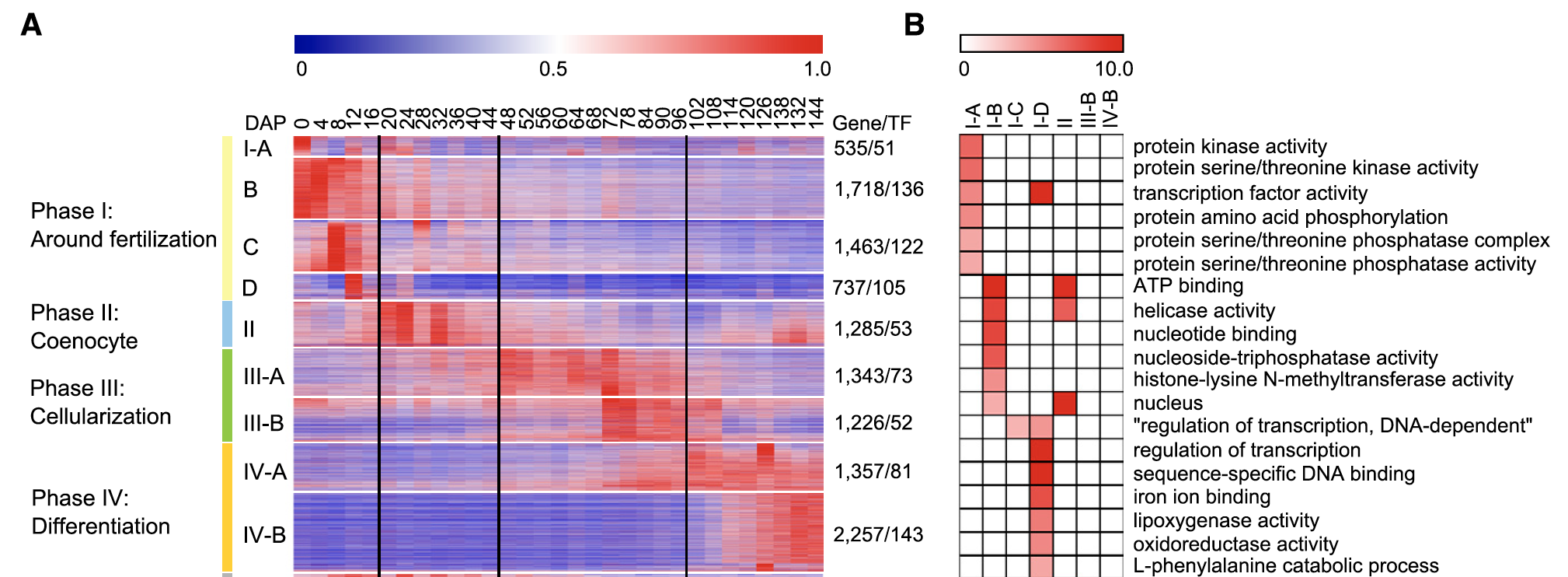
当然,也不能忽略大量基因在多个发育阶段出现,如下:We found that a total of 10,869 genes, including 599 TFs, in modules of M10 ; M18 were expressed at more than one of the four stages
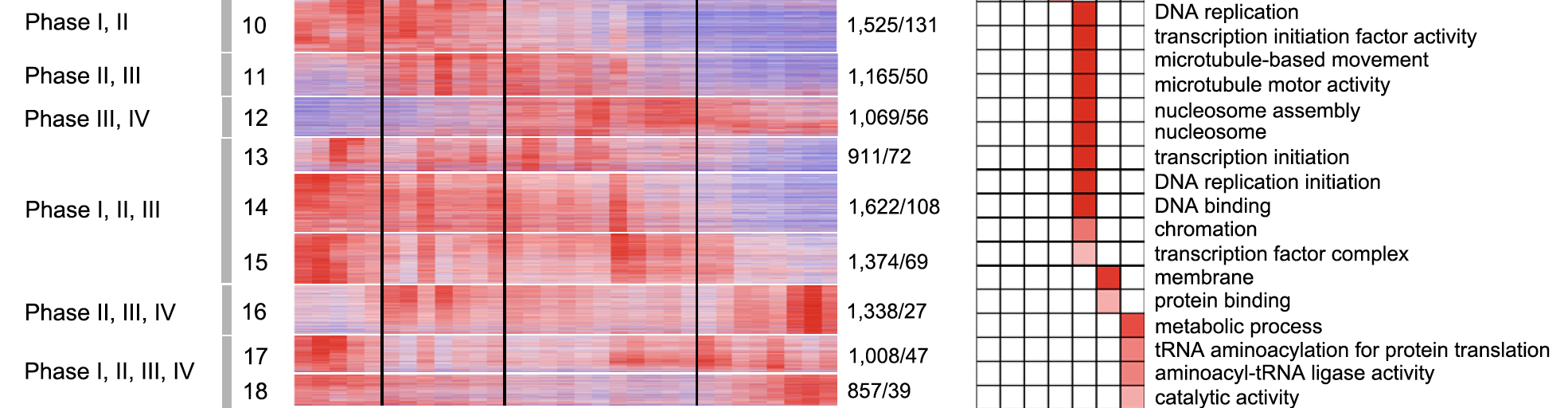
不同的发育阶段会有一些共同的生物学过程。
值得一提的是,因为这里的数据是玉米物种,所以富集分析这里使用MapMan, Mapman是一款老牌但却持续更新的植物基因功能分析,通路分析的软件。做植物分子生物学研究,组学研究的工作者,应是都有所了解。反正我是不了解啦。
Mapman除了对基因进行注释及表达数据可视化之外,还可以利用软件之际对多样品数据进行聚类分析。主要操作过程如下: 从Experiments导入表达数据,具体参考文章:植物代谢通路注释+基因表达可视
文章里面的描述是:
Functional category enrichment for each coexpression module was evaluated with the MapMan (v3.6.0) functional annotation使用MeV软件找基因共表达
TIGR推出的微阵列分析软件包之一。MultiExperiment Viewer的缩写, 通用微阵列分析工具,运用各种算法对格式化好的微阵列数据进行聚类、统计、热图显示、分析。
本研究作者就是采用MeV软件: we clustered all 22,790 expressed genes, including 1415 (6.2%) TFs into 18 coexpression modules using the k-means clustering algorithm。
然后把这18个基因模块一个个描述它们的生物学意义, 其实我们通常是使用WGCNA来做这个分析,我估计作者应该是没有WGCNA的概念,就采用经典而且简单的MeV软件数据下载
其中 RNA-seq data as FPKM values is available through the eFP Browser engine (http://bar.utoronto.ca/efp_maize/cgi-bin/efpWeb.cgi?dataSource=Early_Seed), 如果要下载原始数据,需要服务器去处理,在 PRJNA505095.
作者使用hisat2软件把测序的fastq文件比对到玉米的B73参考基因组,然后用cufflinks计算基因的RPKM值,
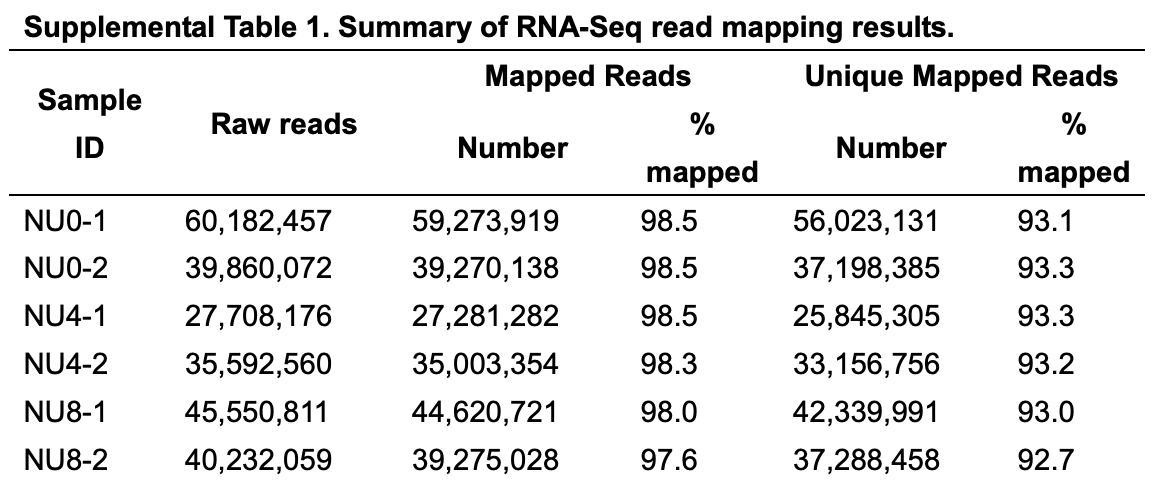
测序数据量不小:
使用公共数据库
作者这里在寻找种子特异性表达基因的时候,使用了公共数据,如下:

调控网络
研究者这里推断gene regulatory network (GRN)的方法参考两个文献

该网络预测了1,317个转录因子和14,540个基因之间的31,256个相互作用。