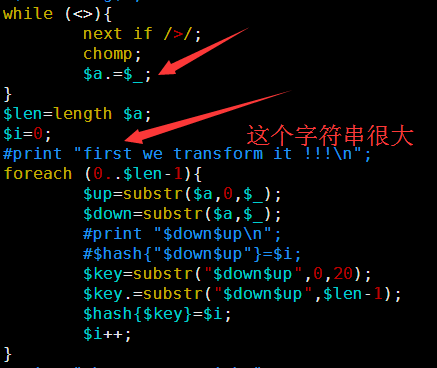
其中第一讲我提到了一个简单的索引产生方式,因为是课堂就半个小时想的,很多细节没有考虑到,对病毒那种几K大小的基因组来说是很简单的,速度也非常快,但是我测试了一下酵母,却发现好几个小时都没有结果,我只好kill掉重新改写算法,我发现之前的测序最大的问题在于没有立即substr函数的实现方式,把一个5M的字符串不停的截取首尾字符串好像是一个非常慢的方式。
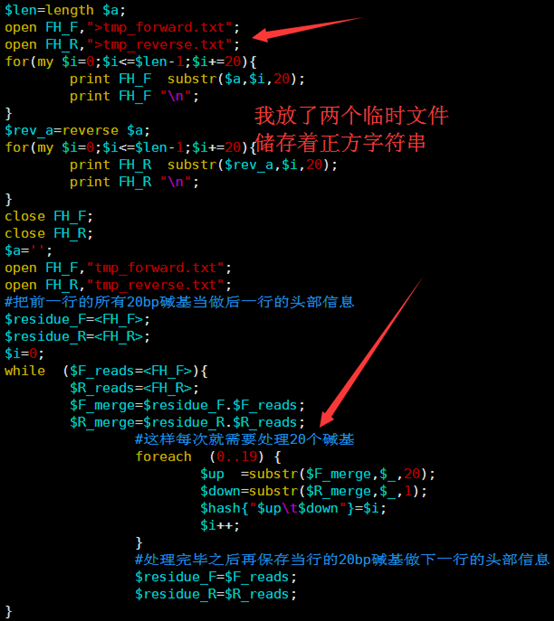
所以我优化了那个字符串的函数,虽然代码量变多了,实现过程也繁琐了一点,但是速度提升了几千倍。
time perl bwt_new_index.pl e-coli.fa >e-coli.index
测试了一下我的脚本,对酵母这样的5M的基因组,索引耗费时间是43秒
real 0m43.071s
user 0m41.277s
sys 0m1.779s
输出的index矩阵如下,我简单的截取头尾各10行给大家看,一点要看懂这个index。
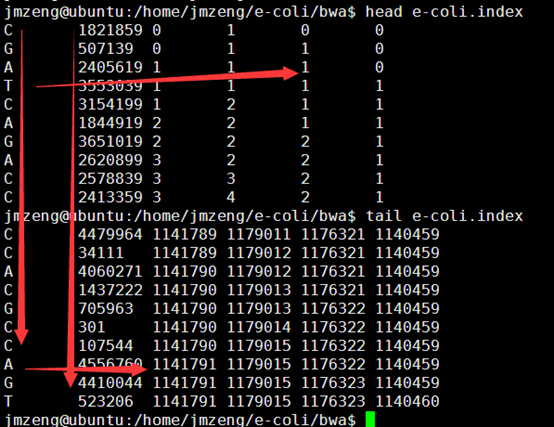
首先第一列就是我们的BWT向量,也就是BWT变换后的尾字符
第二列是之前的顺序被BWT变换后的首字符排序后的打乱的顺序。
第三,四,五,六列分别是A,C,G,T的计数,就是在当行之前累积出现的A,C,G,T的数量,是对第一列的统计。
这个索引文件将会用于下一步的查询,这里贴上我新的索引代码,查询见下一篇文章
[perl]
while (<>){
next if />/;
chomp;
$a.=$_;
}
$len=length $a;
open FH_F,">tmp_forward.txt";
open FH_R,">tmp_reverse.txt";
for(my $i=0;$i<=$len-1;$i+=20){
print FH_F substr($a,$i,20);
print FH_F "\n";
}
$rev_a=reverse $a;
for(my $i=0;$i<=$len-1;$i+=20){
print FH_R substr($rev_a,$i,20);
print FH_R "\n";
}
close FH_F;
close FH_R;
$a='';
open FH_F,"tmp_forward.txt";
open FH_R,"tmp_reverse.txt";
#把前一行的所有20bp碱基当做后一行的头部信息
$residue_F=<FH_F>;
$residue_R=<FH_R>;
$i=0;
while ($F_reads=<FH_F>){
$R_reads=<FH_R>;
$F_merge=$residue_F.$F_reads;
$R_merge=$residue_R.$R_reads;
#这样每次就需要处理20个碱基
foreach (0..19) {
$up =substr($F_merge,$_,20);
$down=substr($R_merge,$_,1);
$hash{"$up\t$down"}=$i;
$i++;
}
#处理完毕之后再保存当行的20bp碱基做下一行的头部信息
$residue_F=$F_reads;
$residue_R=$R_reads;
}
#print "then we sort it\n";
$count_a=0;
$count_c=0;
$count_g=0;
$count_t=0;
foreach (sort keys %hash){
$first=substr($_,0,1);
$len=length;
$last=substr($_,$len-1,1);
#print "$first\t$last\t$hash{$_}\n";
$count_a++ if $last eq 'A';
$count_c++ if $last eq 'C';
$count_g++ if $last eq 'G';
$count_t++ if $last eq 'T';
print "$last\t$hash{$_}\t$count_a\t$count_c\t$count_g\t$count_t\n";
}
unlink("tmp_forward.txt");
unlink("tmp_reverse.txt");
[/perl]