很久以前我们介绍过Sushi这个R包可以绘制基因组区域reads覆盖情况,这次我们介绍另外一个功能更强大的R包 Gviz,全称是Plotting data and annotation information along genomic coordinates 正式发表与期刊 Source Code Biol Med. 2016; 文章链接是 doi: 10.1186/s13029-016-0052-z
官方说明文档是多达200多页的PDF,依赖于很多bioconductor的基础R包,所以这个时候学习这个R包的速度其实取决于我们自己对其它R包或者R基础知识的掌握程度。
说明书地址:http://52.71.54.154/packages/devel/bioc/vignettes/Gviz/inst/doc/Gviz.html

当然我们需要根据示例来学习:
| PDF | R Script | Gviz users guide |
| —————————————————————————————— | —————————————————————————————— | ———————— |
| | | |
重点是 plotTracks 函数,可以根据一系列 GeneRegionTrack 对象进行绘图,包括:

如果你陷入了无穷尽的细节里面,那么你可能需要把那200页的PDF全部读完并且亲自实践一遍,才算是学会,实际上这样并不是好的学习方法,下面就根据我的介绍来逐步掌握它吧!
首先是画染色体示意图
既然我们使用R包 Gviz是为了可视化reads的覆盖情况,那么参考基因组的染色体是必不可少的环节,最简单的展示如下:
library(Gviz)
idTrack <- IdeogramTrack(chromosome=21, genome="hg38")
plotTracks(idTrack, from=500, to=900000)
如果染色体的起始终止坐标跨度比较大, 这个绘图会比较耗时,理论上我们可以展示所有参考基因组的所有染色体的任意起始终止坐标位置情况。
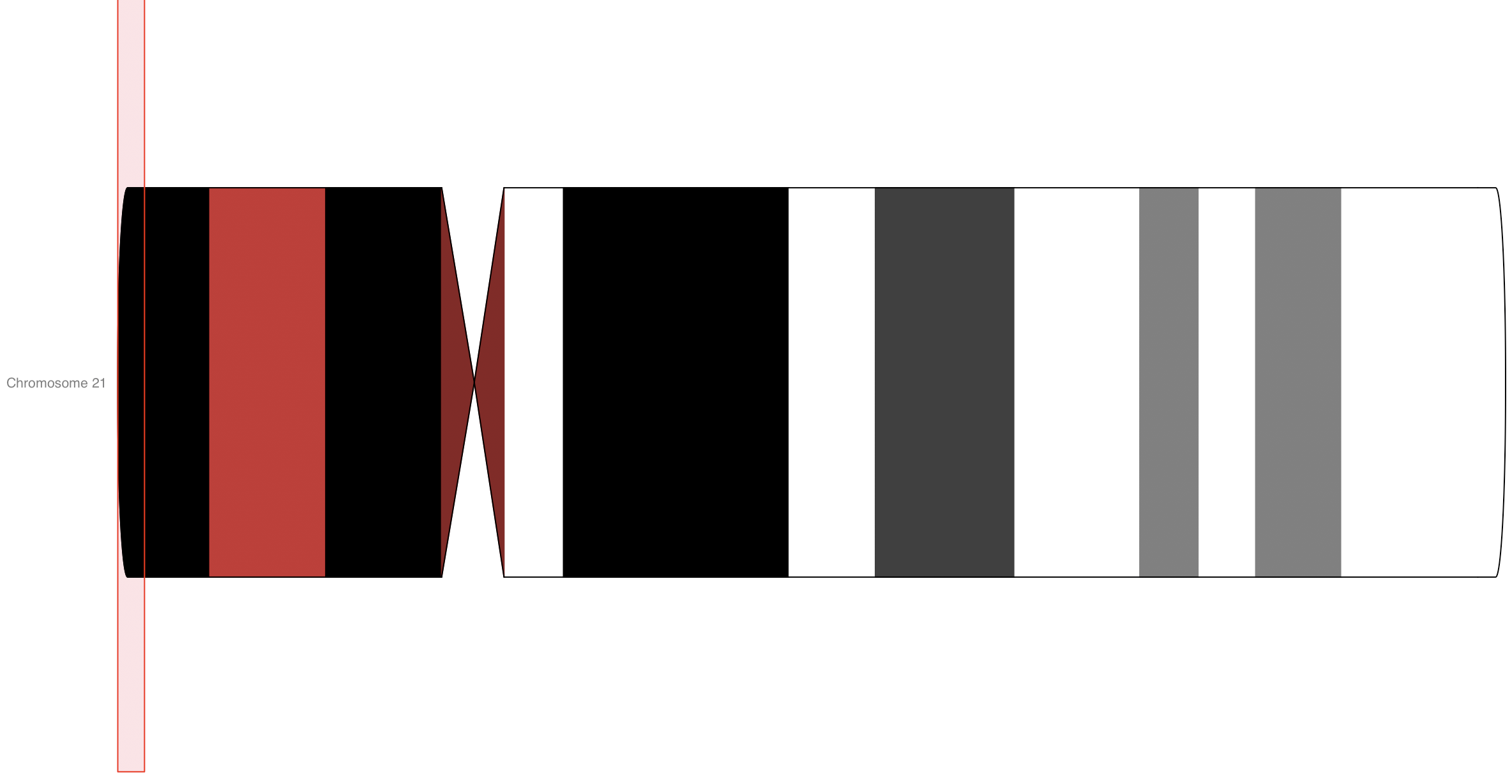
当然,这个用法单独被使用比较少见,通常是作为其它数据的陪衬。
画基因结构
首先要拿到基因的坐标咯,作者文档在这里使用的是基于hg19参考基因的chr17染色体上面的cpg岛的坐标,本质上就是 GRanges 对象加上R包 Gviz 的壳变成 AnnotationTrack 对象。
library(GenomicRanges)
data(cpgIslands)
class(cpgIslands)
chr <- as.character(unique(seqnames(cpgIslands)))
gen <- genome(cpgIslands)
atrack <- AnnotationTrack(cpgIslands, name="CpG")
plotTracks(atrack)
比如我想绘制基因ID是1的那个基因的全部外显子;
library(TxDb.Hsapiens.UCSC.hg38.knownGene)
txdb=TxDb.Hsapiens.UCSC.hg38.knownGene
g1 = exonsBy(txdb)[[1]]
atrack <- AnnotationTrack(g1, name="g1")
plotTracks(atrack)
如下所示,这个基因有3个外显子,实际上仍然是很无聊哦,但是基础知识必须学。

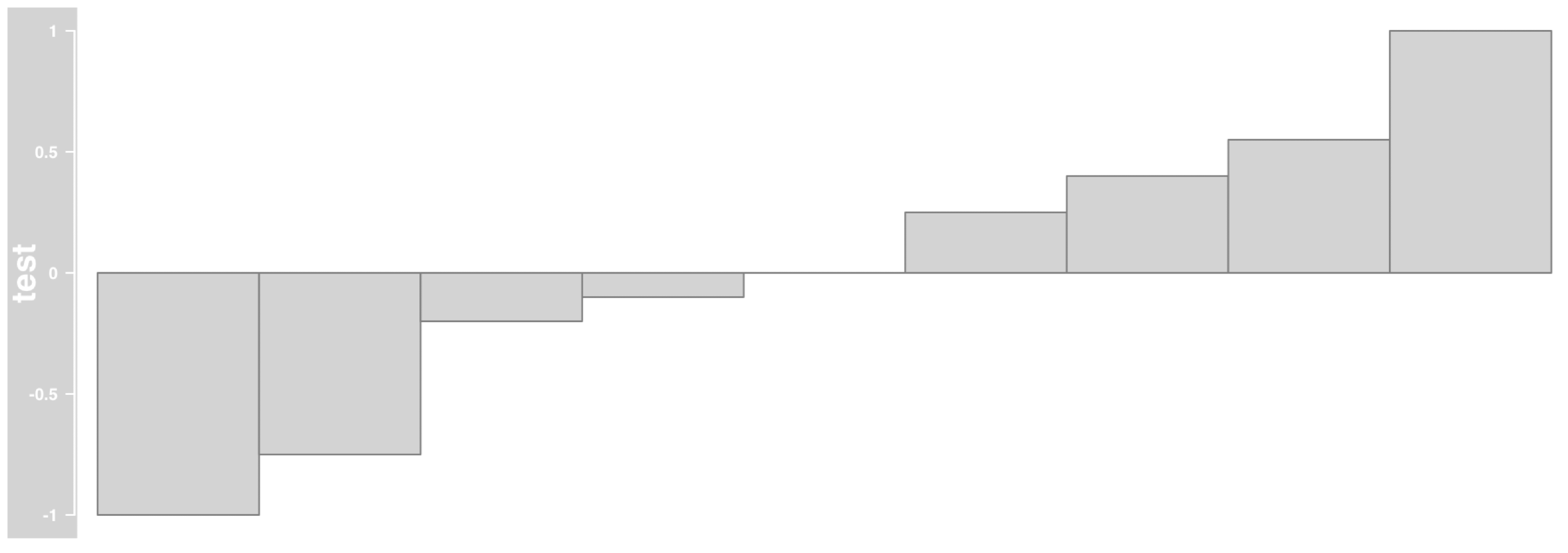
画覆盖度
需要读取bw格式的reads覆盖情况文件,这个时候可以使用R包自带的bw文件,使用 函数 DataTrack 来读取bw文件:
bgFile=system.file("extdata" ,package = 'Gviz',"test.wig")
bw<-DataTrack(range = bgFile,genome="hg19",
type="histogram", name='test')
plotTracks(bw)
大概如下:
把染色体信息,基因结构信息,覆盖度情况结合起来
假设,我们上面3个例子绘制的都是同一个区域的图,就可以结合:
tracklist=list()
tracklist[['bw']]=bw
tracklist[['atrack']]=atrack
tracklist[['idTrack']]=idTrack
plotTracks(tracklist)
其中 bw, atrack, idTrack 都是 AnnotationTrack 对象,它们组合为一个list,就可以一起绘制啦。
实际上,你这个时候看R包的文档,或者IGV,会发现图非常的漂亮,而我们举例的这些,简直是惨不忍睹。
可以这么说,这个R包是可以完全替代IGV的, 前提是你对它的了解足够深。
画其它信息
自行读文档学习,还可以调整多个参数,可以这么说,参数复杂到很多人都不想去看:

R包的示例数据,都可以 载入慢慢玩:
list.files(system.file("data" ,package = 'Gviz'))