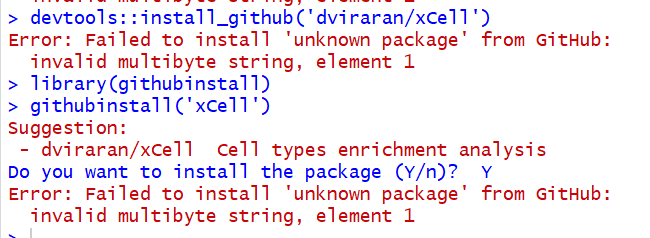
有同学提问,遇到下面的报错,但是网上其他朋友都可以顺利运行,为什么单单是她报错呢?
我觉得这并不是一个好的提问方式,所以用这个例子,来演示如何是一个好的提问。
首先摆出背景知识
第一是吸引大家的注意,感兴趣的就会跟你一起探索,帮助你解决问题,第二个作用是,同时让其他人学到知识,他们或许会投桃报李。
XCELL的背景知识,就是写清楚为什么要用它:
我们都知道,对肿瘤病人进行WES或者WGS等基因组测序后会得到成百上千的somatic突变,而这些突变都是ATCG碱基直接 变化,所以它们变化的组合情况就6种,而加上上下文碱基也就96种,这96种碱基变化的比例的特殊组成,就是mutation signatures,而且COSMIC数据库上面有着30种已知的signatures,我们可以把这些signatures当做是一个有意义的生物学功能,这样一旦我们拿到自己的突变数据, 就可以通过非负矩阵分解的方法把自己的突变数据分解为这30个signatures的组合,那么如果我们拿到的是表达矩阵呢?因为表达矩阵通常是bulk转录组测序,也就是说本来就是肿瘤细胞以及其肿瘤微环境的各种其他细胞组合而成,同理我们应该是可以根据表达量推断出来他们的细胞组分,当然,这个就需要算法上面的突破啦。低通量实验可以用免疫组化,免疫荧光和流式等方法来获得组织免疫细胞组成,而 bulk tumor转录组数据同样可以通过算法轻松得到肿瘤免疫细胞浸润水平。两篇综述系统性的整理和比较了主流算法,包括:
- 基于GSEA的半定量方法
- Deconvolution algorithms(分为partial deconvolution和complete deconvolution)
其中xCell,MCP-counter都是基于ssGSEA所开发出来的方法,CIBERSORT,TIMER,quanTIseq采用反卷积。
现在她就想试试看xCell的效果。然后写清楚自己的探索过程
首先肯定是搜索拿到该包的官网链接,下载方式:
然后就可以打开rstudio,贴代码:devtools::install_github('dviraran/xCell') ## 用法是超级简单 library(xCell) exprMatrix = read.table(expr_file,header=TRUE,row.names=1, as.is=TRUE) xCellAnalysis(exprMatrix)再贴上面那个报错截图!
如果报错,就报告自己的版本信息,比如我的R version 3.5.3 (2019-03-11) -- "Great Truth" Copyright (C) 2019 The R Foundation for Statistical Computing Platform: x86_64-apple-darwin15.6.0 (64-bit)这样帮助其他人快速反应,排查错误!
最后写清楚自己遇到报错后的思考程度
如果你有过思考,就写一下自己的推理过程。
那么她为什么不是这样提问呢?
因为我耗费了15min才写清楚前因后果,我相信大部分人都是不愿意花费这个时间的,所以最佳选择是先丢问题,运气好就有人解答了,如果确实没有人恰好愿意解答,再想办法丰富自己的提问技巧。
另外一个提问该如何修改呢?
有朋友问:
请问用Seurat3.0 pbmc.data <- Read10X载入数据时 出现error:Barcode file missing 怎么办?是不是可以辅助一下截图加上自己的代码呢?