我列过一个生物信息学入门200篇NGS文献解读计划,其中一个文献是发表于2018的NC,标题是:Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq 对6个TNBC病人总共测了 超过1500个单细胞 ,质控后还剩下1189个单细胞进入下游分析。使用的是FACS加上Smart-seq2 ,非常中规中矩的分析,所以就发了同样中规中矩的NC。
组会课题组的博士后分享了一篇nature文章,发表于2019年6月,题目是:CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy 利用了这个数据集加入自己的生物学故事,应该是大家感兴趣的数据挖掘。
该nature文章从公共数据库的原始测序数据开始,自己构建表达矩阵,自己对细胞进行质量控制,自己走单细胞标准流程,就是5个R包,分别是: scater,monocle,Seurat,scran,M3Drop 需要熟练掌握它们的对象,:一些单细胞转录组R包的对象 分析流程也大同小异:
- step1: 创建对象
- step2: 质量控制
- step3: 表达量的标准化和归一化
- step4: 去除干扰因素(多个样本整合)
- step5: 判断重要的基因
- step6: 多种降维算法
- step7: 可视化降维结果
- step8: 多种聚类算法
- step9: 聚类后找每个细胞亚群的标志基因
- step10: 继续分类
文章描述流程如下:

因为原文有标记细胞类群,也有marker,所以作者很容易出图如下:
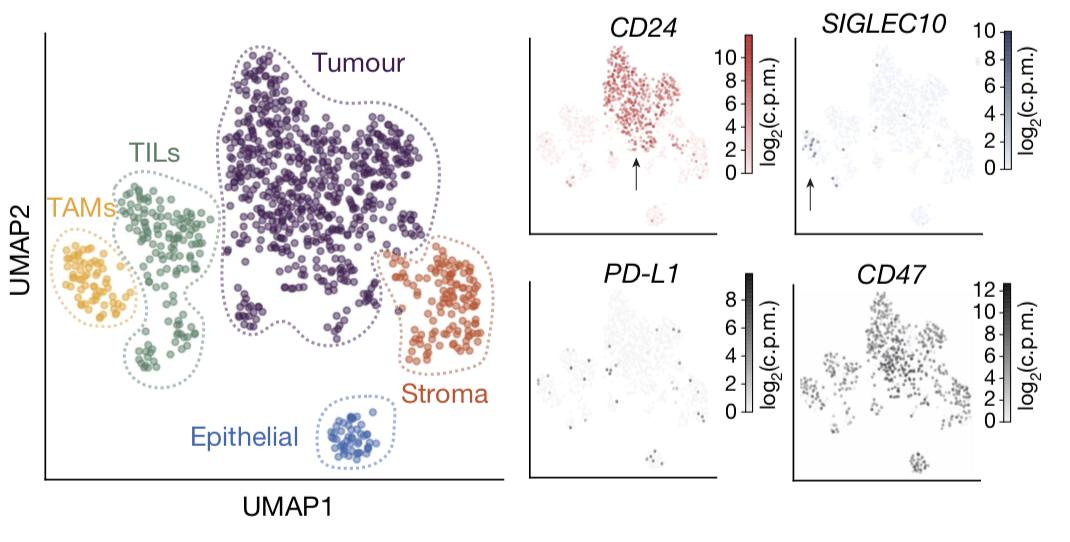
就查看自己感兴趣的基因就好了,毕竟是要使用公共数据库来辅助自己的生物学故事啦。
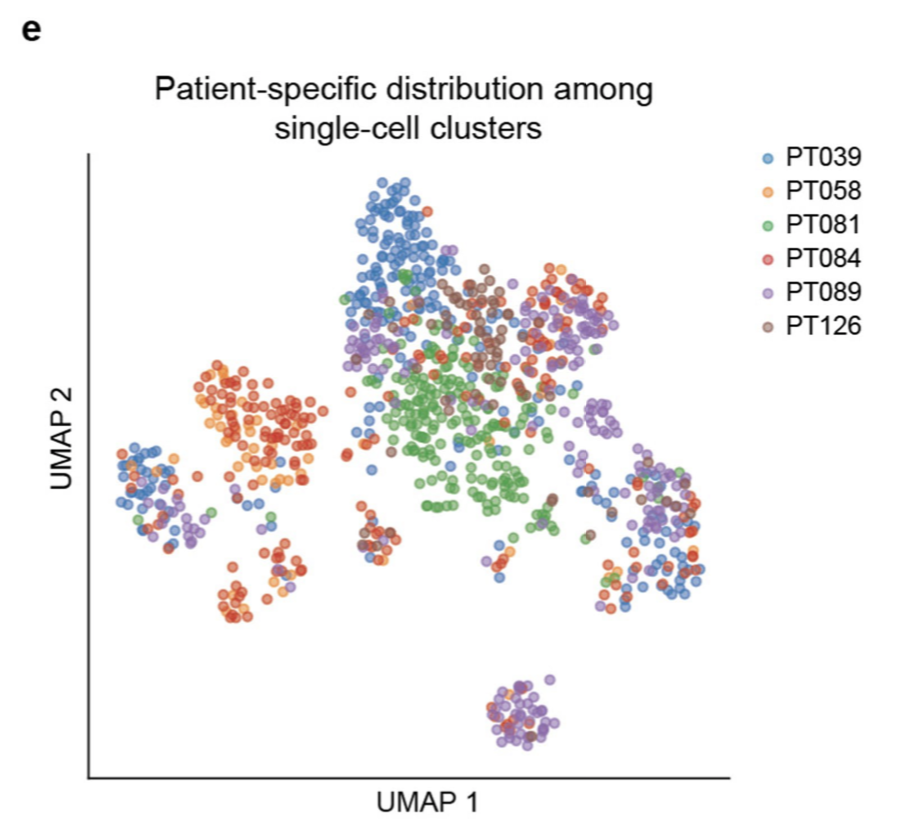
同样的可以看到6个病人,他们的肿瘤细胞是具有病人异质性的,虽然整体是肿瘤细胞,但是病人与病人直接区分的很开,但是其它细胞就是细胞类型特异性很明显,多个病人的同一个细胞类型会被整合在一起。
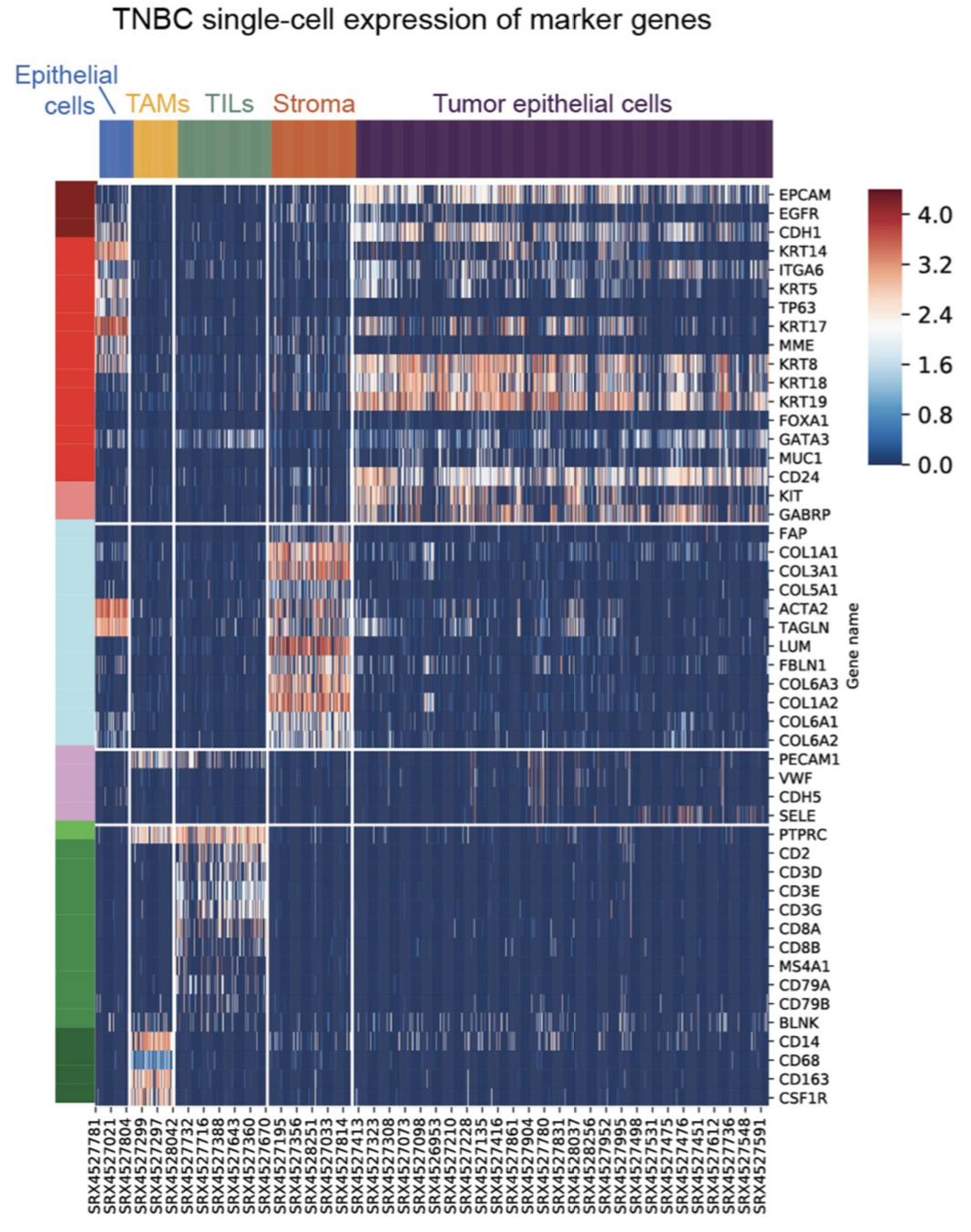
热图展示每个细胞亚群的marker基因也是标配啦
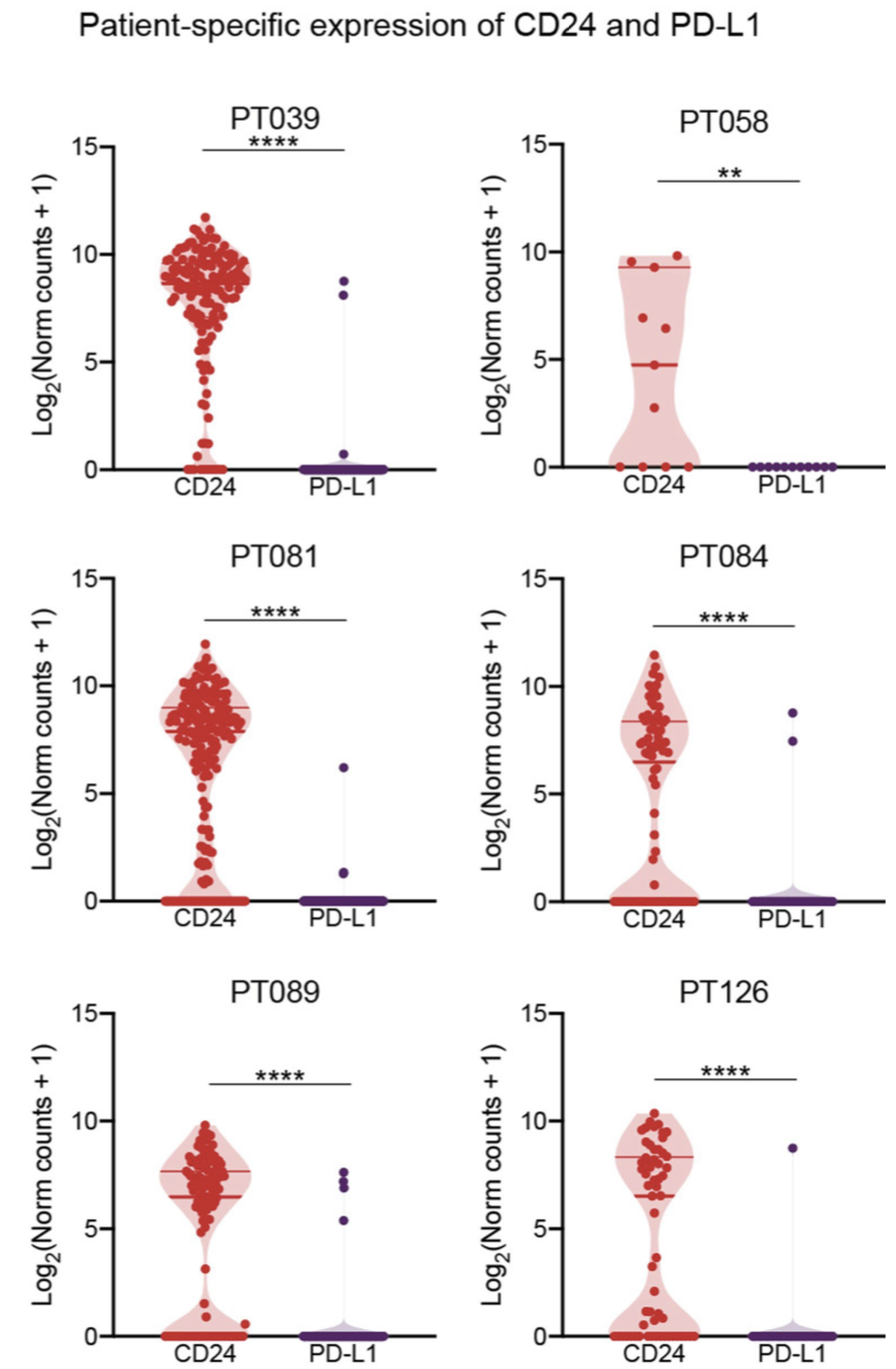
本文不一样的是区分病人来看自己感兴趣的基因的表达情况。
希望这次分享对你的课题有帮助,尝试多看文献,共数据库挖掘需要的基础linux和r技巧好好掌握。