基于fastq测序数据可以做可变剪切,比如bioconductor流程rnaseqDTU 就说明了salmon软件和R包打配合,不过大多数情况下,我们其实已经采用了star或者hisat2软件对fastq测序数据根据参考文件已经进行了比对,这一个步骤非常耗时,所以我们做可变剪切理论上应该是从bam文件开始,省时省力哈!
可变剪切背景知识
可变剪接(Alternative Splicing,AS)是指从一个mRNA前体中通过不同的剪接方式,对外显子和内含子进行组合,产生不同的mRNA剪接异构体的过程。高等真核生物中的可变剪接极大地拓展了基因功能的多样性,是调节基因表达和产生蛋白质组多样性的重要机制。
可变剪接受到具有特殊结构域的顺式调控元件(RNA motif)和识别这些motif的RNA结合蛋白(RNA binding protein,RBP)调控 。RNA-seq通常是二代转录组,可以通过高深度的测序数据组装构建转录本序列,预测外显子与内含子的结构并识别出可变剪接模式,假阳性不小。三代全长转录组利用其读长更长的优势,可以直接读取转录本的全长序列。相比二代测序,无需打断、无需组装,直接获得全长转录本的结构信息,可以更准确的分析生物体内存在可变剪接事件。
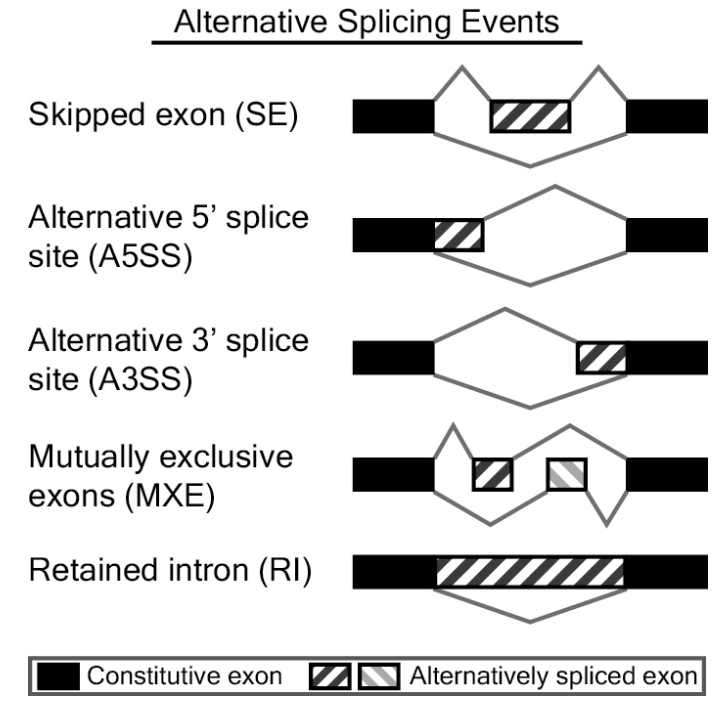
Multivariate Analysis of Transcript Splicing (MATS)软件列出5种可变剪切,分别是skipped exon (SE)外显子跳跃,alternative 5′ splice site (A5SS)第一个外显子可变剪切,alternative 3′ splice site (A3SS)最后一个外显子可变剪切,mutually exclusive exons (MXE)外显子选择性跳跃和 retained intron (RI)内含子滞留,示意图在官网,如下:
SplicSeq软件可以拿到7种可变剪切形式信息:
- 可变受体位点(AA)
- 可变供体位点(AD)
- 可变启动子(AP)
- 可变终止子(AT)
- 内含子保留(RI)
- 外显子跳跃(ES)
- 外显子互斥(ME)
如果你看TCGA的ISOexpresso数据库:http://wiki.tgilab.org/ISOexpresso/ 上面有8种:
可变剪切在癌症领域研究例子
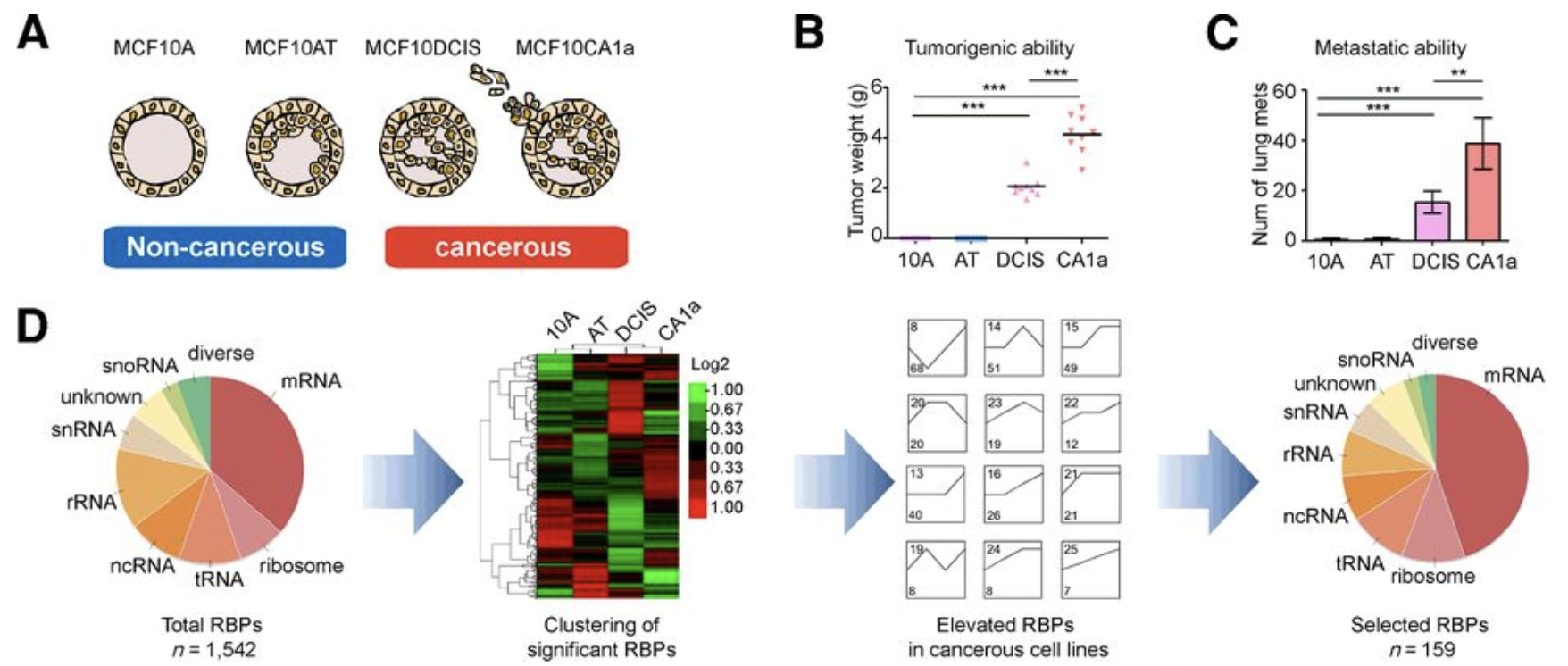
可变剪切在癌症领域研究不多,但其重要性毋庸置疑,主要是因为技术手段限制,值得介绍的经典文章,比如邵志敏教授的题为“PHF5A Epigenetically Inhibits Apoptosis to Promote Breast Cancer Progression.”利用CRISPR-Cas9文库技术对RNA结合蛋白进行系统性功能筛选,发现了乳腺癌生存依赖的的剪切因子PHF5A . TCGA、METABRIC、KMplot以及本中心的研究队列显示,PHF5A在乳腺癌组织中普遍存在高表达,并且其高表达指示着较差的预后。生物学功能上,PHF5A缺失会导致癌细胞增殖、迁移和成瘤能力显著受损。 该文章的测序数据在 SRP139147 ,然后走RNA-seq分析流程,比较cancer cells (CA1a or DCIS) 和noncancer cells (MCF10AT or MCF10A)差异分析,虽然作者文章主要是关心那1,542 manually curated RBPs ( RNA-binding proteins) ,不过最后落脚点仍然是可变剪切:
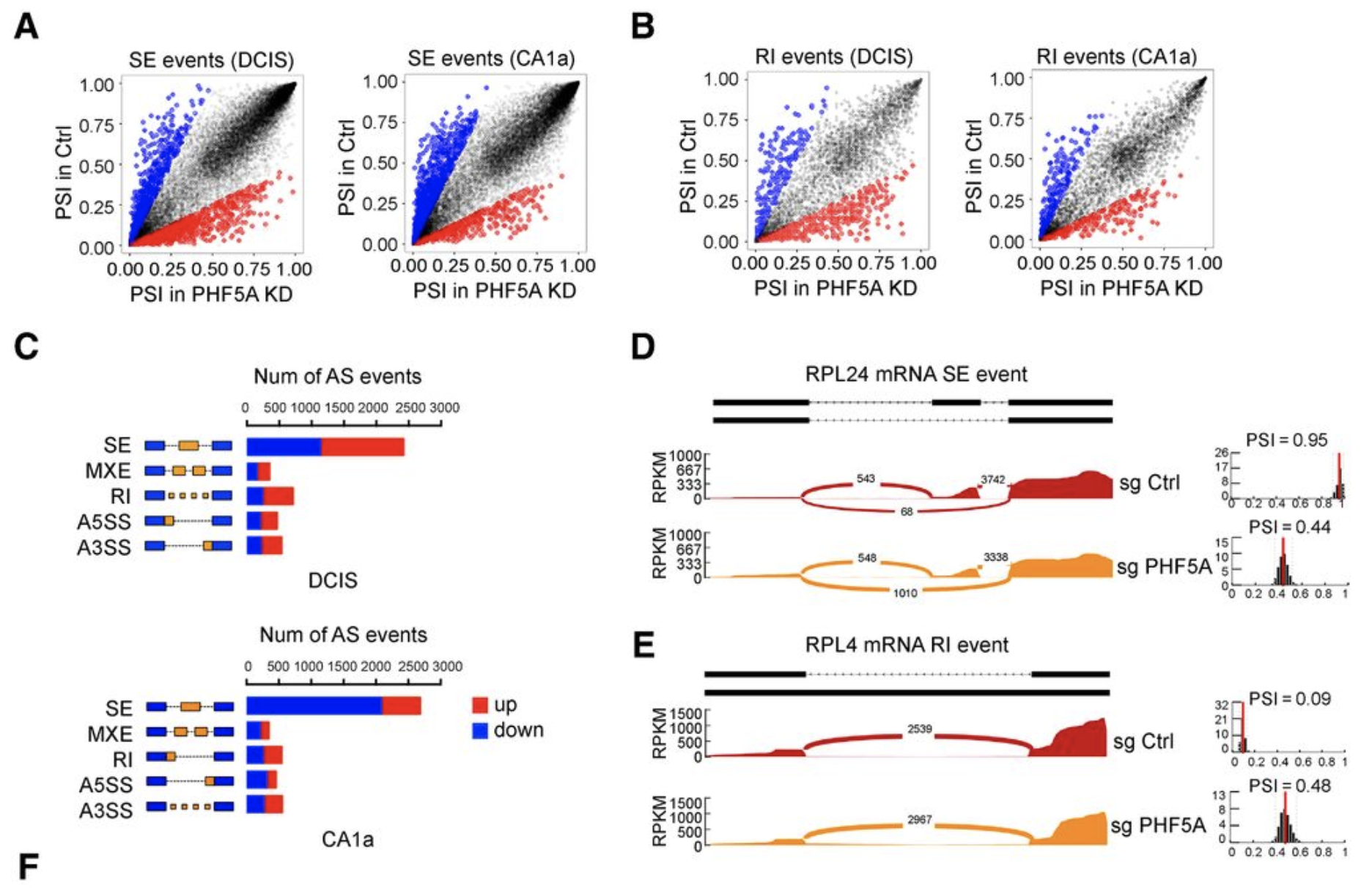
文章写作落脚点是PHF5A knockdown appeared to switch full-length FASTK (FASTK-L) to an intron 5-retained variant (herein termed FASTK short, FASTK-S) in CA1a cells 是通过差异分析筛选出来的。
这个时候,有两个非常经典的软件leafcutter和rMATS,我都在生信技能树写过教程,两年前过去了,现在又需要重新使用,是时候更新一下软件和用法了。
首先看软件各自的官网安装
leafcutter 软件相关地址:
- LeafCutter software, https://github.com/davidaknowles/leafcutter;
- Full documentation is available at http://davidaknowles.github.io/leafcutter/;
- LeafViz visualizations, https://leafcutter.shinyapps.io/leafviz/;
rMATS 软件相关地址:
- rMATS paper:https://www.pnas.org/content/111/51/E5593
- Software Download Release: rMATS 4.0.2 download (Linux or MacOS), includes transcript annotation
- Documentation : User Tutorial
- rmats2sashimiplot for converting the rMATS output into sashimiplot.
- rMAPS: RNA Map Analysis and Plotting Server for the analysis of RNA-binding proteins (RBPs) binding site around the rMATS SE events.
全部使用conda一键式安装吧:
conda create -n isoform
conda activate isoform
conda install -y -c bioconda samtools
conda install -y -c bioconda rmats
conda install -y -c davidaknowles r-leafcutter
因为conda可以自动帮你解决全部的依赖:
比如 r-leafcutter就依赖于一大波的R包,而且leafcutter的GitHub源代码里面有一些脚本和测试数据,所以还是要下载看看
mkdir -p ~/biosoft
cd ~/biosoft
rm -rf leafcutter
git clone https://github.com/davidaknowles/leafcutter
cd leafcutter
## 需要修改里面的一个脚本 scripts/bam2junc.sh 把软件路径增添进去即可
再次吐槽一下,这个软件包里面涵盖了,perl,python,r,sh代码,开发流程非常的不统一!
然后看看软件各自的安装和使用
首先让我们先回顾一下leafcutter 软件的4个标准步骤:
第一个步骤是shell脚本bam2junc.sh把bam文件转为junc文件,可以构建好bam_path.txt文件,存储全部的bam文件路径然后批量处理,第一个步骤全部的bam文件输出的junc文件路径保存在 all_juncfiles.txt 。
第二个步骤是使用python脚本 leafcutter_cluster.py做 Intron clustering,就是把第一个步骤全部的bam文件输出的junc文件合并得到_perind_numers.counts.gz 文件,里面每一行都是一个内含子,每一列都是一个样本,写明了它们的表达值,这些数值就可以用来做可变剪切分析。
ls ~/data/public/star/*bam > bam_path.txt
cat bam_path.txt |while read id
do
file=$(basename $id )
sample=${file%%.*}
echo Converting $id to $sample.junc
sh ~/biosoft/leafcutter/scripts/bam2junc.sh $id $sample.junc
done
ls *.junc >all_juncfiles.txt
# 需要python2.7版本
conda activate isoform
python ~/biosoft/leafcutter/clustering/leafcutter_cluster.py -j all_juncfiles.txt -m 50 -o chinese_tnbc -l 500000
第3个步骤是使用R脚本 leafcutter_ds.R :对整个项目的样本进行分组,然后对它们的tumor_perind_numers.counts.gz 文件分类计算差异,其中物种的指定参考基因组版本的外显子坐标文件需要自己制作,比如软件自带的:gencode19_exons.txt.gz 格式如下所示:
chr start end strand gene_name
chr1 11869 12227 + DDX11L1
chr1 12613 12721 + DDX11L1
chr1 13221 14409 + DDX11L1
自己写脚本制作,比如:
zcat ~/biosoft/leafcutter/leafcutter/data/gencode19_exons.txt.gz|head
wget ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_32/gencode.v32.annotation.gtf.gz
zcat gencode.v32.annotation.gtf.gz |perl -alne '{next unless $F[2] eq "exon";/gene_name \"(.*?)\"/;print "$F[0]\t$F[3]\t$F[4]\t$F[6]\t$1"}' > ~/biosoft/leafcutter/leafcutter/data/gencode_v32_hg38_exons.txt
然后,分组文件 group_info.txt 也需要自己制作,是两列的格式, 样本名和分组,举例如下:
SRR2016934 control
SRR2016948 control
SRR2016953 control
SRR2016965 control
SRR2016932 npc
SRR2016933 npc
SRR2016935 npc
SRR2016936 npc
SRR2016937 npc
SRR2016938 npc
运行第三步代码如下:
exonbed=/home/yb77613/biosoft/leafcutter/leafcutter/data/gencode_v32_hg38_exons.txt
head $exonbed
conda activate isoform
~/biosoft/leafcutter/scripts/leafcutter_ds.R --num_threads 4 \
--exon_file=$exonbed --min_samples_per_intron=4 \
tumor_perind_numers.counts.gz group_info.txt
第4个步骤是使用R脚本 ds_plots.R 对差异分析结果进行可视化。
exonbed=/home/yb77613/biosoft/leafcutter/leafcutter/data/gencode_v32_hg38_exons.txt
head $exonbed
conda activate isoform
~/biosoft/leafcutter/scripts/ds_plots.R -e $exonbed \
tumor_perind_numers.counts.gz group_info.txt \
leafcutter_ds_cluster_significance.txt -f 0.05
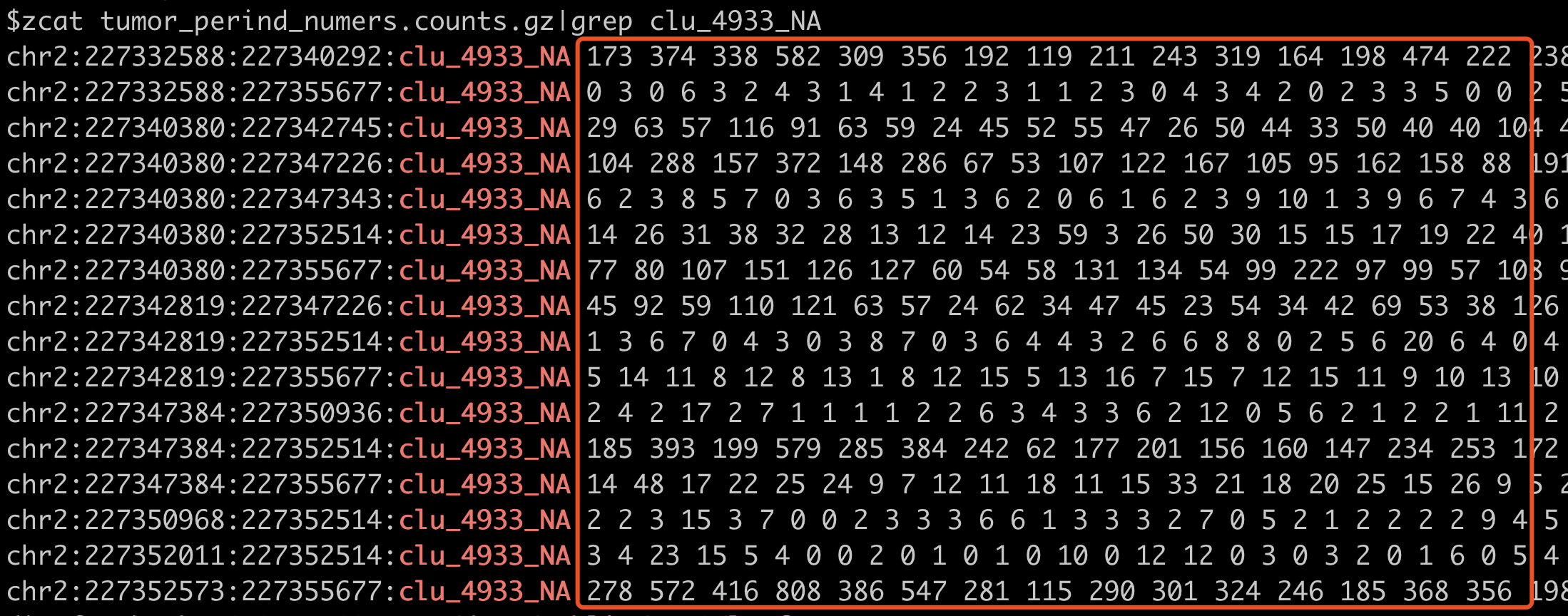
所有的统计学显著的可变剪切形式都会可视化在一张图里面,也可以自己别出心裁的使用其它可视化方法。我们随机展示其中一个结果(MFF基因):

chr2:clu_4932_NA Success 4.6012973874349 7 0.238436066745941 0.634806885002018 MFF
chr2:clu_4933_NA Success 81.7113181603927 15 5.08675778183616e-27 1.39918539586235e-23 MFF
看外显子表达量是:
软件就是根据分组,做一下统计检验看看是否有差异。
接着让我们看看rMATS v4.0.2 (turbo)的用法
它只有一个步骤,直接根据分组对bam文件进行可变剪切的差异分析,通过统计模型对不同样本(有生物学重复的)进行可变剪切事件的表达定量,然后以likelihood-ratio test计算P value来表示两组样品在IncLevel(Inclusion Level)水平上的差异(从公式上来看,IncLevel跟PSI的定义也是类似的),lncLevel并利用Benjamini Hochberg算法对p value进行校正得FDR值。
gtf="/home/yb77613/reference/gtf/gencode/gencode.v32.annotation.gtf"
rmats.py --b1 control.bam.txt --b2 tumor.bam.txt \
--gtf $gtf --nthread 5 \
--od bam_test -t paired --readLength 100
主要运行过程中,是一些python模块,或者系统库文件缺失会报错,比如我遇到的:
Running the statistical part.
/home/yb77613/miniconda3/envs/isoform/rMATS/rMATS_C/rMATSexe: error while loading shared libraries: libgsl.so.0: cannot open shared object file: No such file or directory
Traceback (most recent call last):
File "/home/yb77613/miniconda3/envs/isoform/rMATS/rMATS_P/FDR.py", line 45, in <module>
ifile=open(sys.argv[1]);title=ifile.readline();
IOError: [Errno 2] No such file or directory: '/tmp/tmphKCv6b/JC_SE/rMATS_result_P-V.txt'
paste: /tmp/tmphKCv6b/JC_SE/rMATS_result_FDR.txt: No such file or directory
搜索了一些解决方案,最后采纳:
echo $LD_LIBRARY_PATH
LD_LIBRARY_PATH=/home/yb77613/miniconda3/envs/isoform/lib/
echo $LD_LIBRARY_PATH
我们运行的rMATS 程序首先会解析gtf文件:
There are 60609 distinct gene ID in the gtf file
There are 227462 distinct transcript ID in the gtf file
There are 36694 one-transcript genes in the gtf file
There are 1372308 exons in the gtf file
There are 25234 one-exon transcripts in the gtf file
There are 22480 one-transcript genes with only one exon in the transcript
Average number of transcripts per gene is 3.752941
Average number of exons per transcript is 6.033131
Average number of exons per transcript excluding one-exon tx is 6.661165
Average number of gene per geneGroup is 8.466432
然后读取全部的bam文件。
Done processing each gene from dictionary to compile AS events
Found 48974 exon skipping events
Found 4028 exon MX events
Found 16561 alt SS events
There are 10052 alt 3 SS events and 6509 alt 5 SS events.
Found 6969 RI events
但是,很明显,仍然是报错的, 因为图便利,采用了conda安装,代码是:conda install -y -c bioconda rmats
可以看到,conda虽然说能解决初学者99%的软件安装问题,但也不是万能的。
比较两个分析的结果
rMATS运行失败,懒得去解决它的bugs了,以后再说。
和salmon加DRIMSeq流程比较
前面我们介绍过,不需要走bam这个文件格式做中间产物,在Swimming downstream: statistical analysis of differential transcript usage following Salmon quantification 流程里面也可以进行转录本差异分析
可变剪切的结果,其实就是转录本差异分析,所以也可以互相比较的。比如前面的MFF基因,搜索:
gene_id "ENSG00000168958.19"; transcript_id "ENST00000456345.1"; gene_type "protein_coding"; gene_name "MFF";
对应的转录本ID,发现也是差异的:
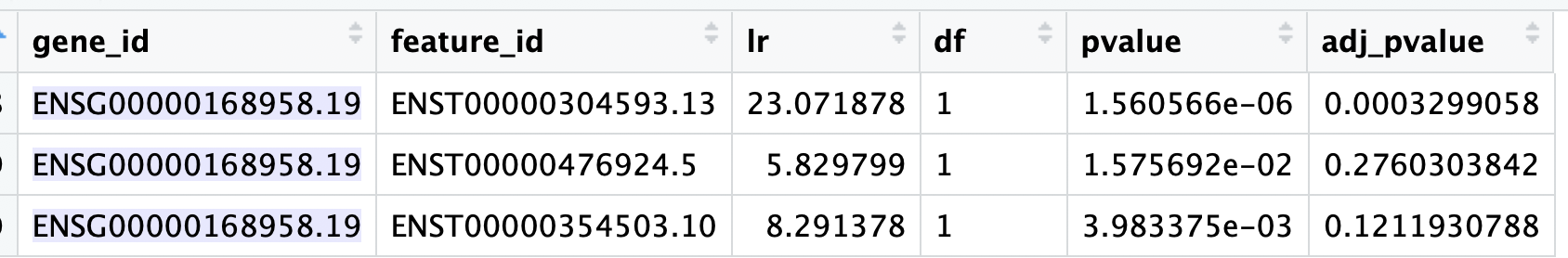
这个基因的3个差异转录本,可视化如下:
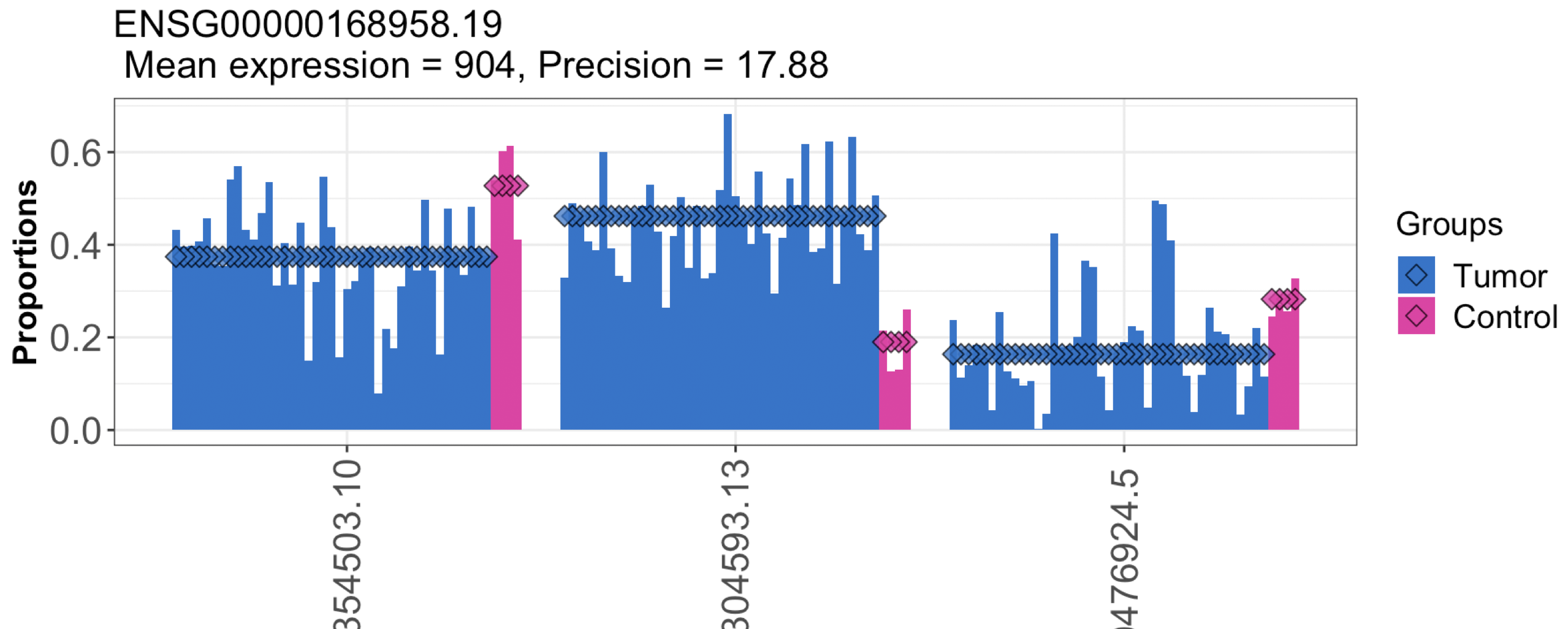
可以看到,寻找差异转录本,或者差异外显子的分析思路,是基于已有的注释,在人类这个物种的研究当然是非常顺畅的。
既然不同的算法,拿到的结果大同小异,我们可以放心的走下一步啦!