我们多次在生信技能树公众号介绍过star-fusion这个目前最好的针对RNA-seq测序数据找融合基因的软件:最好用的融合基因查找工具终于正式发表了 ,还有一个踩过的坑需要注意:[一个好像没有做任何改变的参数](https://mp.weixin.qq.com/s?__biz=MzAxMDkxODM1Ng==&mid=2247491737&idx=2&sn=f61dfe578cbc13635518222e559810be&scene=21#wechat_redirect) 但是关于融合基因的后续生物学介绍我们说的不够,现在就带领大家仔细理解一下star-fusion软件的结果!
我们的示例项目得到的结果,按照JunctionReadCount排序如下:
#FusionName JunctionReadCount SpanningFragCount
FGFR3--TACC3 773 1087
IGL-@-ext--PRAMENP 216 1343
IGL-@--PRAMENP 216 1343
IPO7--LINC02489 186 1724
PPFIA1--TSGA10IP 141 213
RNF121--COBL 125 6
IGL-@-ext--AL121832.3 121 0
IGL-@--AL121832.3 121 0
IGL-@-ext--FLOT2 108 1
IGL-@--FLOT2 108 1
按照SpanningFragCount排序如下:
IPO7--LINC02489 186 1724
IGL-@-ext--PRAMENP 216 1343
IGL-@--PRAMENP 216 1343
FGFR3--TACC3 773 1087
NFKB2--AC025062.3 82 770
IGL-@-ext--PRAMENP 25 366
IGL-@--PRAMENP 25 366
FOXP1--IGL@-ext 9 345
FOXP1--IGL@-ext 28 345
FOXP1--IGL@-ext 27 345
其中FGFR3—TACC3这个融合基因事件我们比较熟悉了,就拿它为例子讲解如何理解这个融合现象, 首先查看具体信息:
FGFR3--TACC3 773 1087 ONLY_REF_SPLICE
FGFR3^ENSG00000068078.18 chr4:1806934:+
TACC3^ENSG00000013810.19 chr4:1735731:+
YES_LDAS 33.6089 GT 1.8892 AG 1.9656
# 下面的是FusionAnnotator (bundled with STAR-Fusion)注释结果的整合
["Cosmic","ChimerPub","YOSHIHARA_TCGA","TumorFusionsNAR2018",
"ChimerKB","ChimerSeq","CCLE_StarF2019","TCGA_StarF2019",
"GUO2018CR_TCGA","Klijn_CellLines","INTRACHROMOSOMAL[chr4:0.05Mb]",
"LOCAL_REARRANGEMENT:+:[48117]"]
可以看到4号染色体的两个临近基因的融合
首先IGV可视化
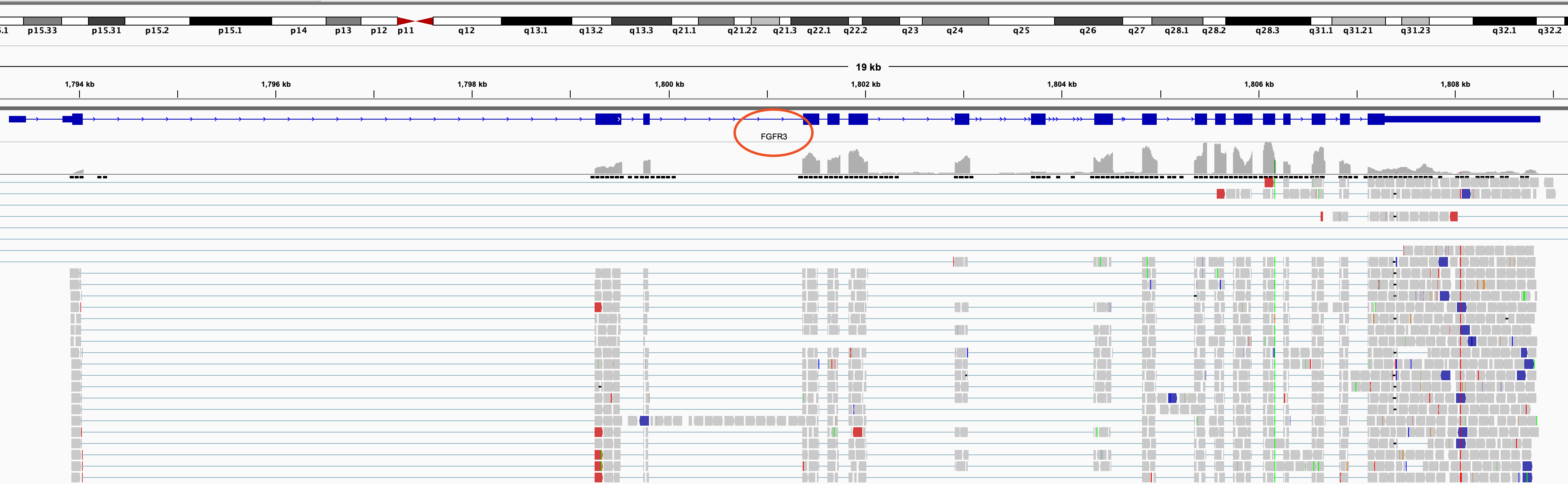
得到的结果通常是需要可视化,如果我们单独的IGV可视化FGFR3基因如下:
如果我们单独的IGV可视化TACC3基因如下:
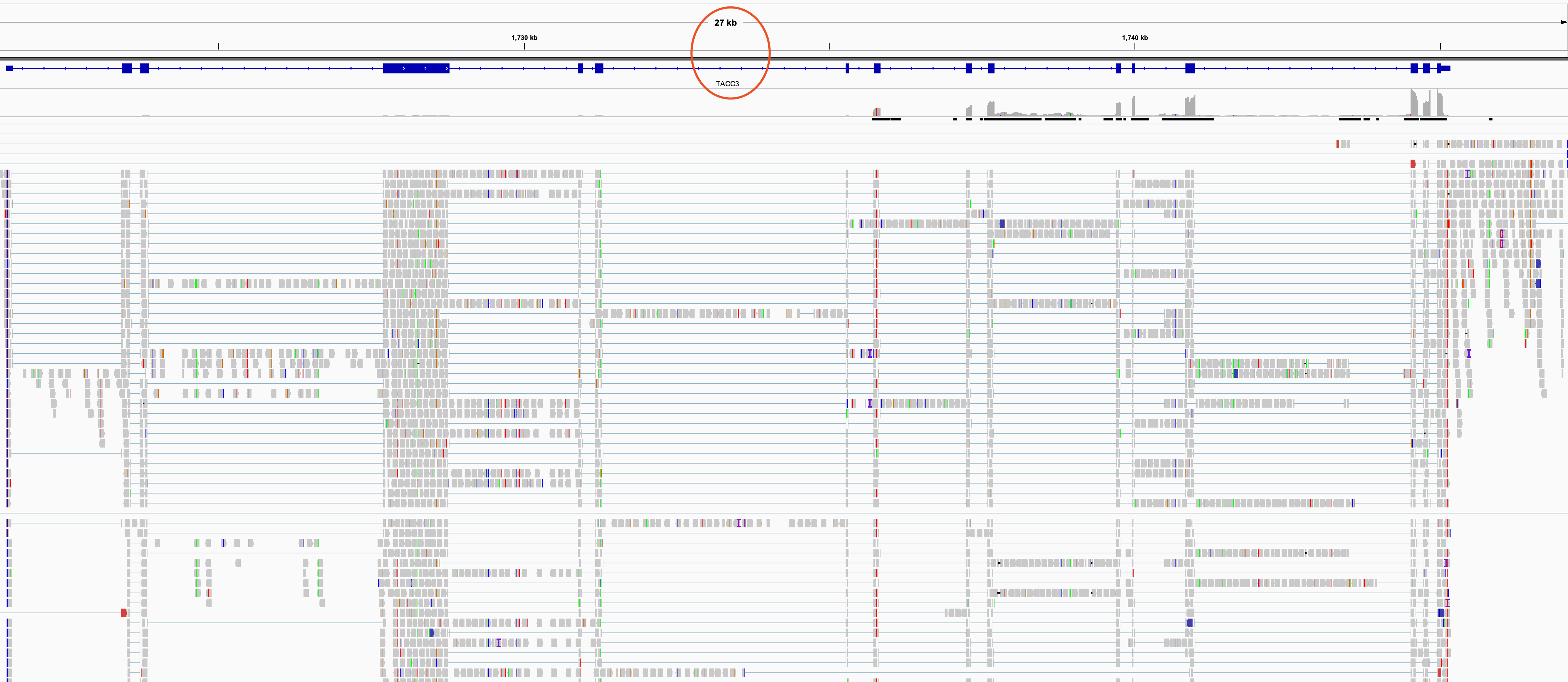
可以看到这两个基因都有点长, 我们的NGS测序也就100bp罢了,它们两个基因都是几十个Kb长度。所以这样是无法浏览融合事件的,需要采用高级工具,比如FusionInspector
然后使用FusionInspector仔细检查
它虽然说是star-fusion御用小工具,但不限于star-fusion的融合事件的检查,可以是其它一系列软件出来的融合基因结果,包括: Prada, FusionCatcher, SoapFuse, TophatFusion, DISCASM/GMAP-Fusion, STAR-Fusion, or other), 的检查。因为它的输入数据要求很简单,就是两个基因的融合关系,如下:
B3GNT1--NPSR1
ZNF709--DYRK1A
ZNF844--NCBP2
RBX1--HAPLN2
FAM180B--TRIM60
CASP9--ADCYAP1
HS3ST3A1--C1QTNF2
OPTC--AP000347.4
GRIA2--ZW10
运行FusionInspector工具:
conda install -c bioconda fusion-inspector
bin_star='~/biosoft/STAR-2.7.3a/bin/Linux_x86_64/STAR'
lib='~/biosoft/starFusion/db/GRCh38_gencode_v31_CTAT_lib_Oct012019.plug-n-play/ctat_genome_lib_build_dir/'
FusionInspector --fusions SRR2016940_fusionList.txt \
-O SRR2016940
--genome_lib $lib \
--left_fq ~/clean/SRR2016940_1_val_1.fq.gz --right_fq ~/clean/SRR2016940_2_val_2.fq.gz \
--out_prefix finspector \
--vis
FusionInspector工具其实是一个打包好的流程,中间过程太多,而且大多数是perl代码,就不一一介绍了,其中最后居然是使用 create_fusion_report.py 这个python代码出报告。如果你没有安装 https://github.com/FusionInspector/fusion-reports 工具,就会报错:
cd ~/miniconda3/envs/rna/share/fusion-inspector-2.2.1-0/fusion-reports/
git clone https://github.com/FusionInspector/fusion-reports.git
pip install igv_reports
~/miniconda3/envs/rna/share/fusion-inspector-2.2.1-0/fusion-reports/create_fusion_report.py \
--html_template ~/miniconda3/envs/rna/share/fusion-inspector-2.2.1-0/util/fusion_report_html_template/igvjs_fusion.html \
--fusions_json ~/fusion/FI/finspector.fusion_inspector_web.json \
--input_file_prefix finspector \
--html_output ~/fusion/FI/finspector.fusion_inspector_web.html
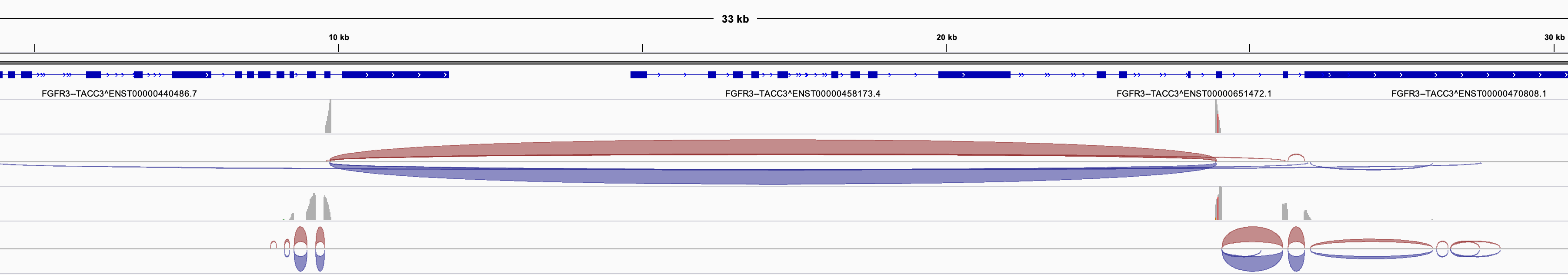
没有html报告问题不大,我们可以把数据文件载入到IGV自己进行可视化,需要下面的4个文件:
finspector.fa : the candidate fusion-gene contigs (if you copy things elsewhere, make sure to als copy the index file finspector.fa.fai)
finspector.bed : the reference gene structure annotations for fusion partners
finspector.junction_reads.bam : alignments of the breakpoint-junction supporting reads.
finspector.spanning_reads.bam : alignments of the breakpoint-spanning paired-end reads.
载入IGV即可,当然了,这个时候需要对IGV有一点认识才行!
如果要批量检验全部样本的star-fusion结果呢
就需要写自动化批量处理的脚本了。这个很难哦,感觉写出来了能看懂的也不多,就算了吧!需要认真学好linux,参考我在 《生信分析人员如何系统入门Linux(2019更新版)》 设定的linux入门6个阶段 ,一般来说,每个阶段都需要至少一天以上的学习:
- 第1阶段:把linux系统玩得跟Windows或者MacOS那样的桌面操作系统一样顺畅,主要目的就是去可视化,熟悉黑白命令行界面,可以仅仅以键盘交互模式完成常规文件夹及文件管理工作。
- 第2阶段:做到文本文件的表格化处理,类似于以键盘交互模式完成Excel表格的排序、计数、筛选、去冗余,查找,切割,替换,合并,补齐,熟练掌握awk,sed,grep这文本处理的三驾马车。
- 第3阶段:元字符,通配符及shell中的各种扩展,从此linux操作不在神秘!
- 第4阶段:高级目录管理:软硬链接,绝对路径和相对路径,环境变量
- 第5阶段:任务提交及批处理,脚本编写解放你的双手
- 第6阶段:软件安装及conda管理,让linux系统实用性放飞自我
最后友情宣传生信技能树
- 生物信息学“义诊”
- 生物信息学”拍卖会”
- 全国巡讲:R基础,Linux基础和RNA-seq实战演练 : 预告:12月28-30长沙站
- 广州珠江新城GEO数据挖掘滚动开班
- DNA及RNA甲基化数据分析与课题设计