最近接到TCGA分析需求,想看看指定基因在指定癌症是否具有临床意义(也就是生存分析是否有统计学显著效果咯!)其实很早以前我在生信技能树就号召粉丝讨论过这个问题:集思广益-生存分析可以随心所欲根据表达量分组吗 这里我做实力演绎一下。
我这里选择最方便的 网页工具:https://xenabrowser.net/heatmap/ 选择合适的数据集及样本信息还有基因来演示一下,随便选择一个基因一个癌症吧,如下:
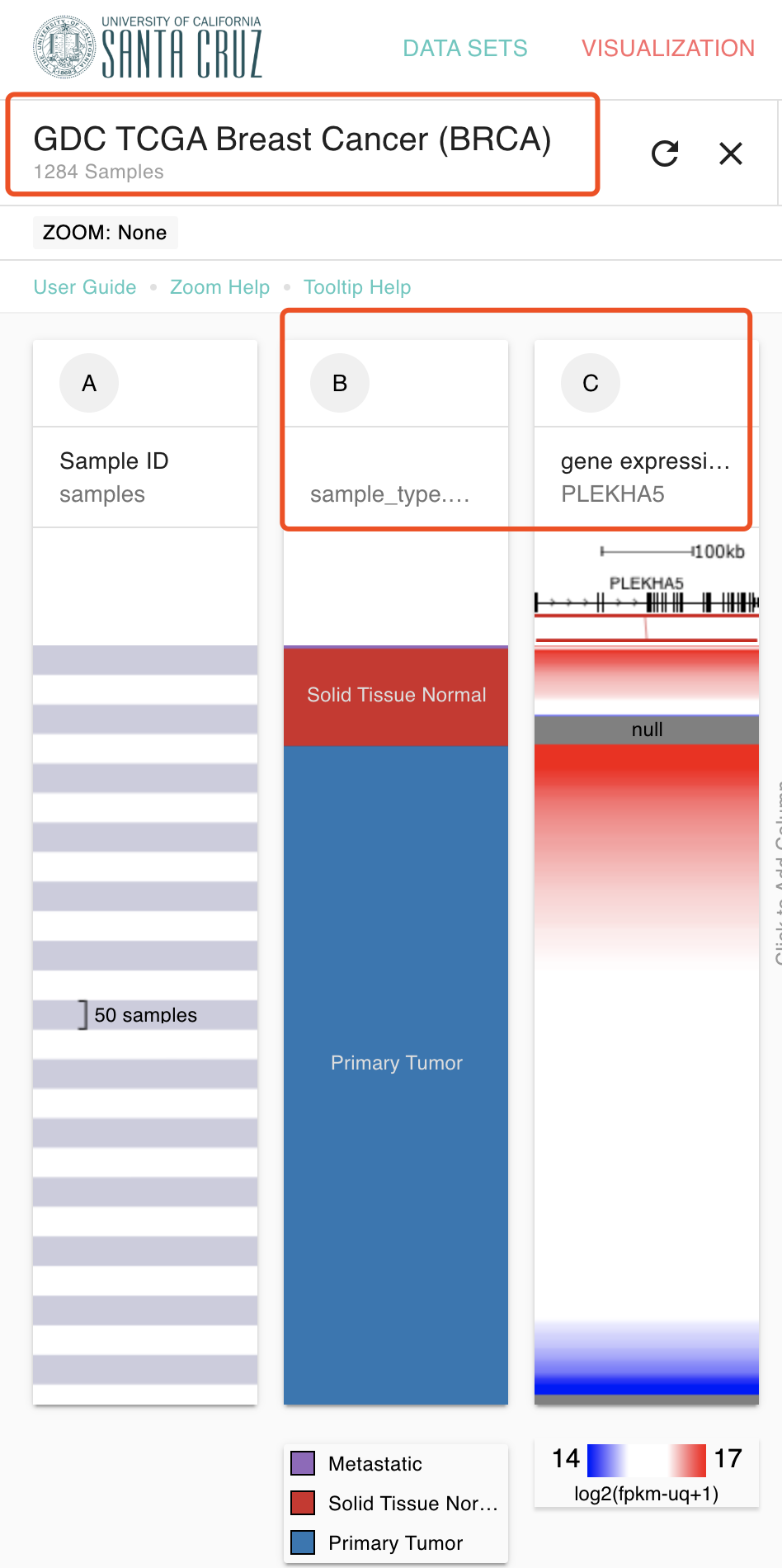
这个时候,我草率的制作了生存分析图如下:
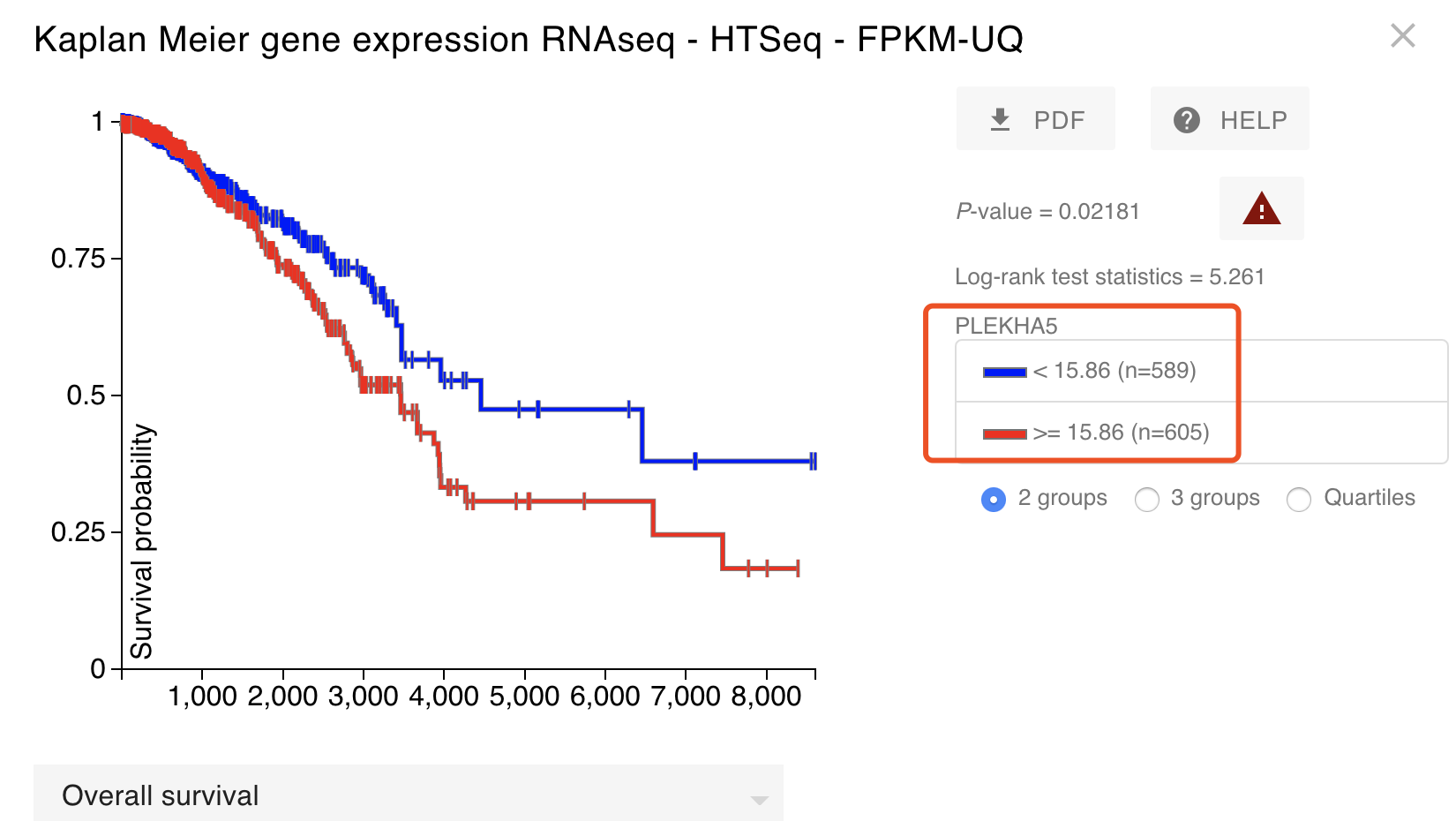
(@ο@) 哇~非常显著,差点准备交差了,然后下意识的看了看病人数量,TCGA数据库的BRCA病人没道理居然快1200个了,肯定是有什么地方错误了,重新看了看,的确是因为没有顾虑到里面有正常组织测序的那些病人,怎么说呢,相当于把有正常组织测序的那一百多个病人,在我这个生存分析里面计算了两次,他们的生存时间信息,生存状态都重复计算了,所以实际上这个生存分析是错误的。
过滤一下,仅仅是保留tumor的表达量信息和病人临床信息,再次制作生存分析曲线,如下所示:
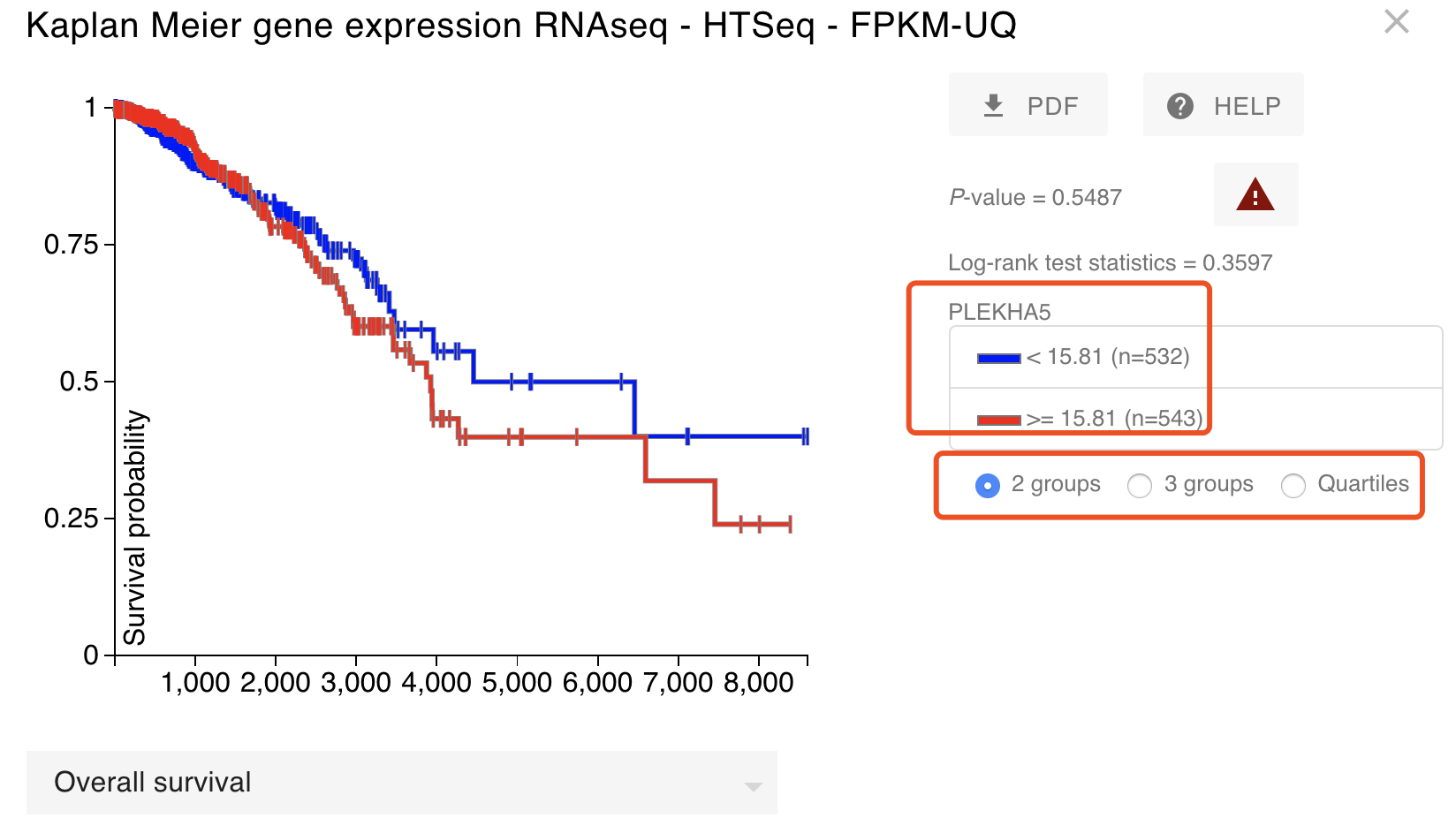
可以看到,之前明明是显著的结果消失了,而且不管是使用哪种表达量划分方式,都达不到统计学显著阈值。
是不是就没有办法了呢?
当然不是,还可以使用R包,一个非常棒的外国小哥博客写的很清楚:http://r-addict.com/2016/11/21/Optimal-Cutpoint-maxstat.html
还有专门的文章,这里就不细心讲解啦。
使用survminer包的surv_cutpoint函数找寻最近生存分析阈值
外国小哥博客写的很清楚:http://r-addict.com/2016/11/21/Optimal-Cutpoint-maxstat.html 我们现在就测试一下这个流程。
首先下载我们前面的数据文件:'PLEKHA5-BRCA.tsv' 内容如下:
总共6列,在前面的 网页工具:https://xenabrowser.net/heatmap/ 选择对应的信息下载即可:
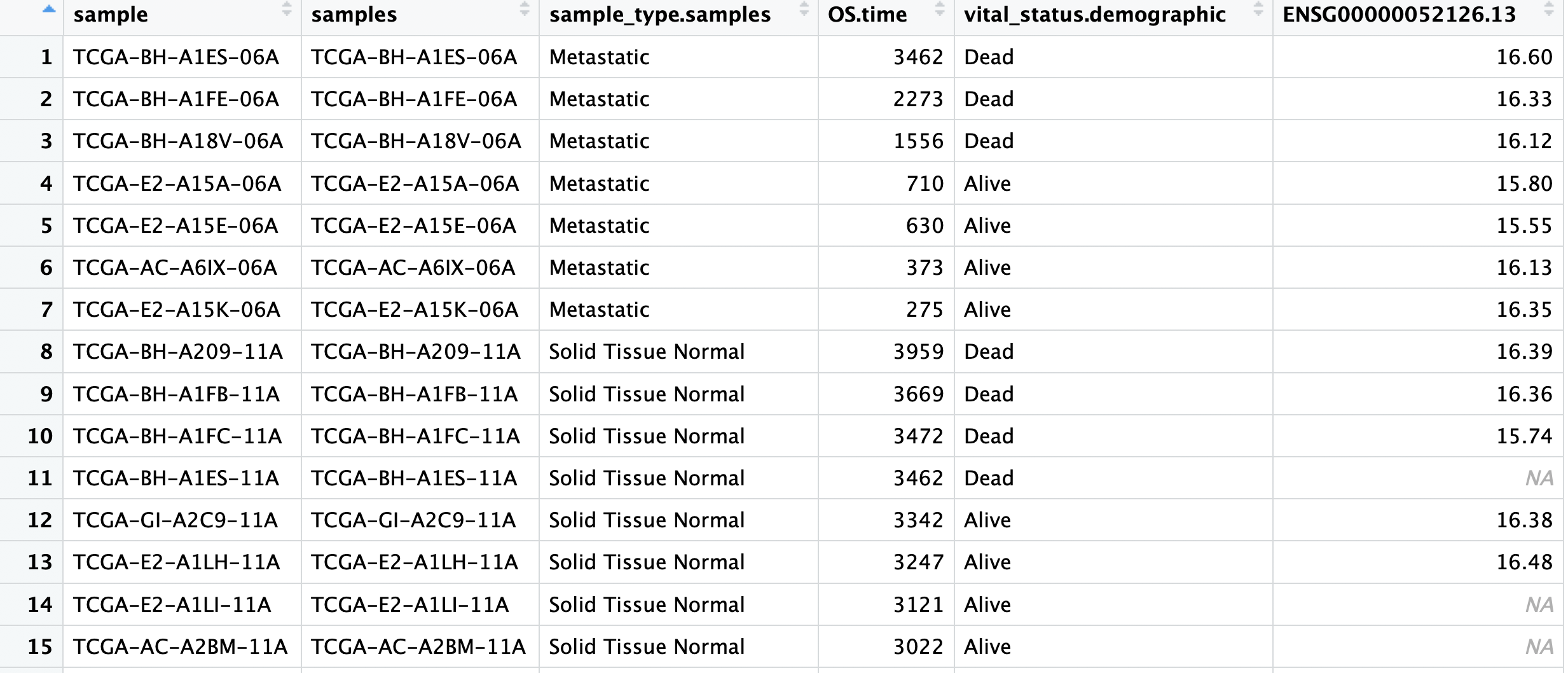
然后是R代码读入上面的文件,主要是列名需要保证正确无误!!!
rm(list=ls())
options(stringsAsFactors = F)
# install.packages("survminer")
library(survminer)
a=read.table('PLEKHA5-BRCA.tsv',header = T,sep = '\t')
head(a)
dat=a[a$sample_type.samples=='Primary Tumor',4:6]
head(dat)
dat$vital_status.demographic=ifelse(dat$vital_status.demographic=='Alive',1,0)
surv_rnaseq.cut <- surv_cutpoint(
dat,
time = "OS.time",
event = "vital_status.demographic",
variables = c("ENSG00000052126.13")
)
summary(surv_rnaseq.cut)
plot(surv_rnaseq.cut, "ENSG00000052126.13", palette = "npg")
surv_rnaseq.cat <- surv_categorize(surv_rnaseq.cut)
library(survival)
fit <- survfit(Surv(OS.time, vital_status.demographic) ~ ENSG00000052126.13,
data = surv_rnaseq.cat)
ggsurvplot(
fit, # survfit object with calculated statistics.
risk.table = TRUE, # show risk table.
pval = TRUE, # show p-value of log-rank test.
conf.int = TRUE, # show confidence intervals for
# point estimaes of survival curves.
xlim = c(0,3000), # present narrower X axis, but not affect
# survival estimates.
break.time.by = 1000, # break X axis in time intervals by 500.
risk.table.y.text.col = T, # colour risk table text annotations.
risk.table.y.text = FALSE # show bars instead of names in text annotations
# in legend of risk table
)
重要的的列名是:
time = "OS.time",
event = "vital_status.demographic",
variables = c("ENSG00000052126.13")
如果是你自己的数据集,需要稍微修改哦。
见证奇迹的时刻:
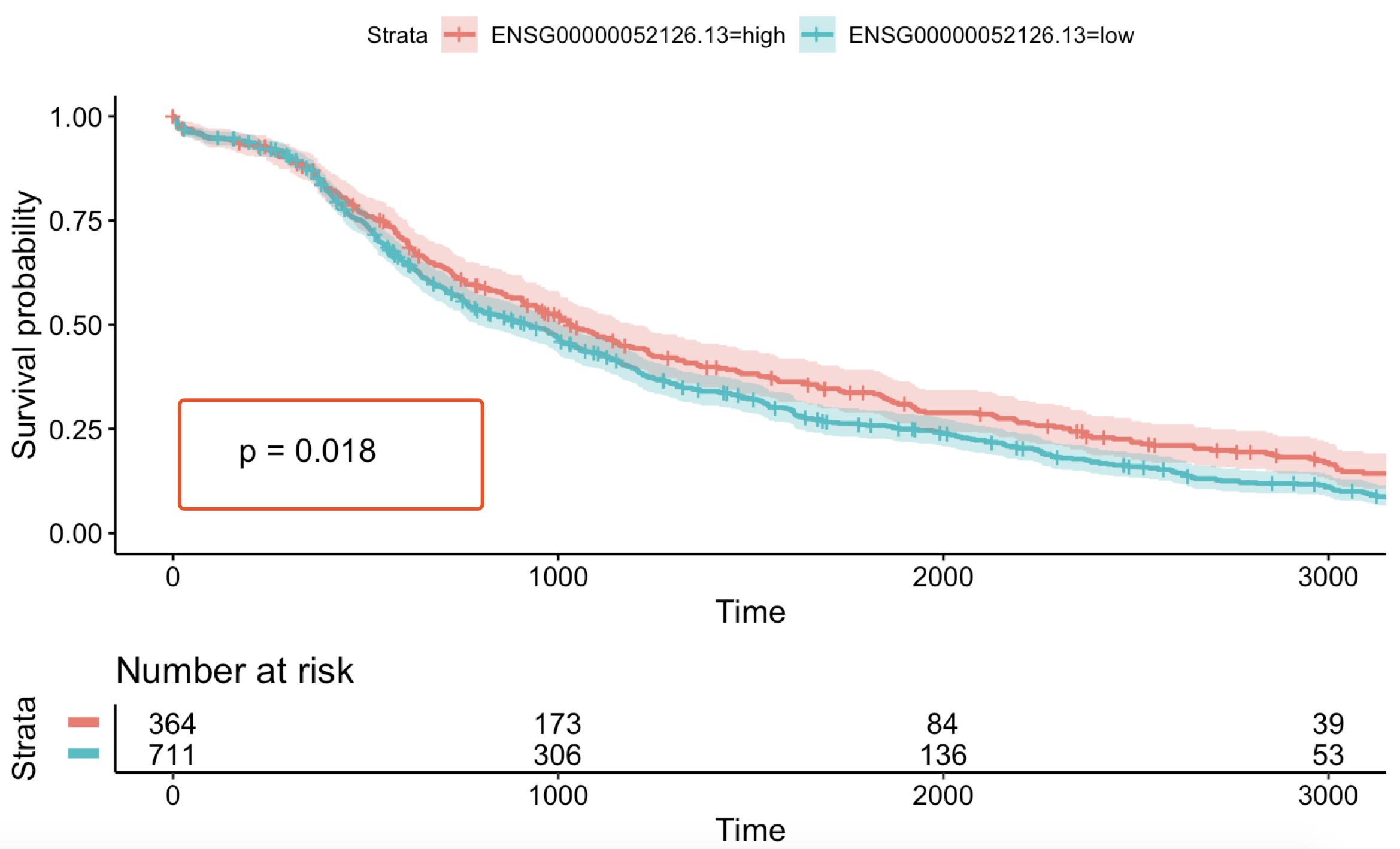
是不是统计学显著啦!!!
函数帮我们选择的分组;
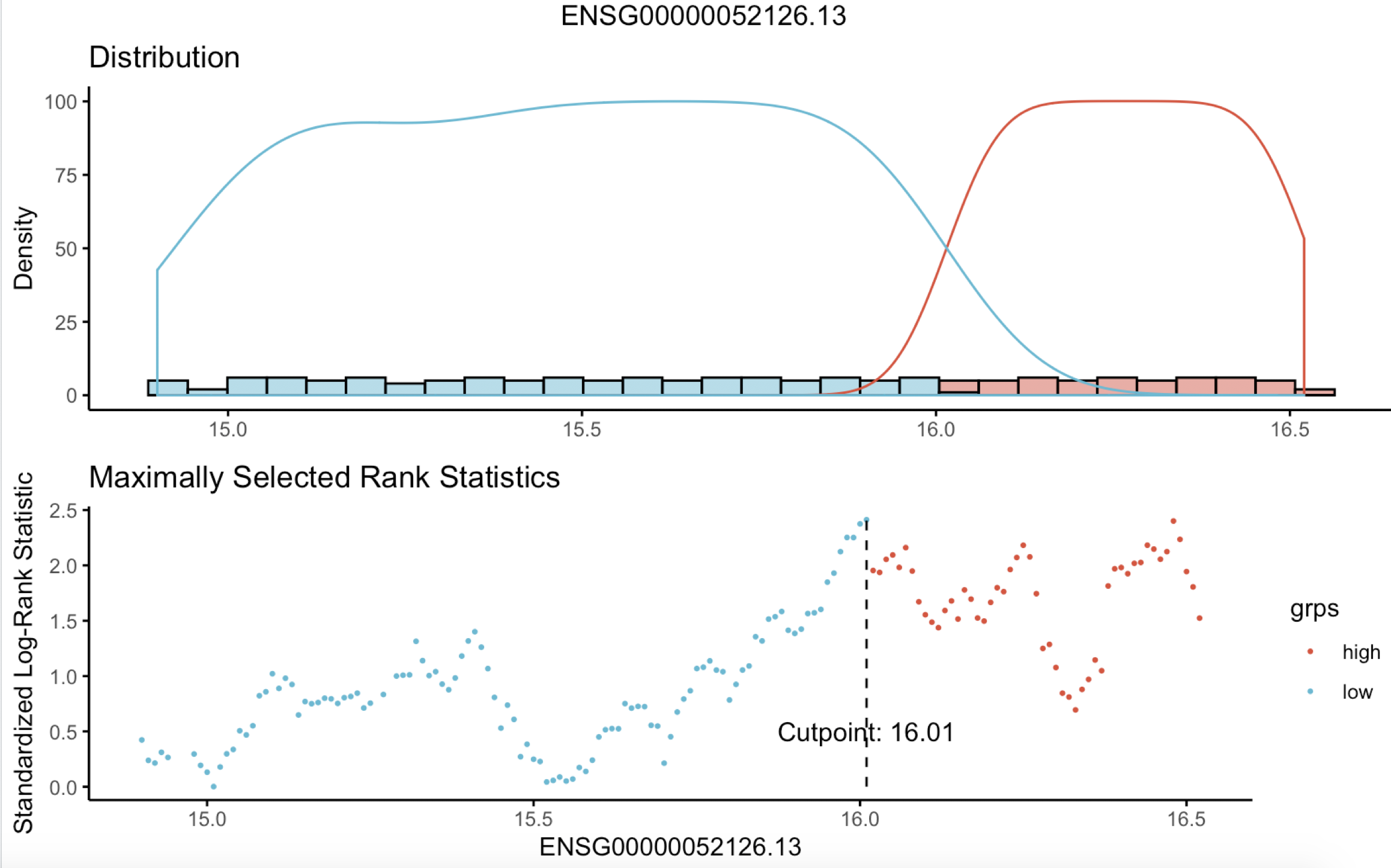
更多TCGA教程见;
- 使用R语言的cgdsr包获取TCGA数据(cBioPortal)
- TCGA的28篇教程- 使用R语言的RTCGA包获取TCGA数据 (离线打包版本)
- TCGA的28篇教程- 使用R语言的RTCGAToolbox包获取TCGA数据 (FireBrowse portal)
- TCGA的28篇教程- 批量下载TCGA所有数据 ( UCSC的 XENA)
- TCGA的28篇教程- 数据下载就到此为止吧
- TCGA的28篇教程- 指定癌症查看感兴趣基因的表达量
- TCGA的28篇教程- 对TCGA数据库的任意癌症中任意基因做生存分析
- TCGA的28篇教程-整理GDC下载的xml格式的临床资料
- TCGA的28篇教程-风险因子关联图-一个价值1000但是迟到的答案
- TCGA的28篇教程-数据挖掘三板斧之ceRNA
- TCGA的28篇教程-所有癌症的突变全景图
- TCGA的28篇教程-早期泛癌研究
- TCGA的28篇教程-CNV全攻略