朋友圈医务工作者不少,经常看到各个疾病方向的义诊通知,各大城市均有,很佩服大家,而且我表示实名羡慕。虽然我不是学医的,但是也可以从另外一个层面帮助一下大家,我也来一个义诊,那就是我们生信技能树最擅长生物信息学方面的“义诊”啦!
大家都知道,因为深度依托于生物学,所以生物信息学也是复杂的不要不要的,比如目前最综合最全面的癌症病人相关组学数据库,TCGA数据库就至少包含8种数据:
- DNA Sequencing
- miRNA Sequencing
- Protein Expression array
- mRNA Sequencing
- Total RNA Sequencing
- Array-based Expression
- DNA Methylation array
- Copy Number array
大部分科研课题组其实做不到每种数据都精通,更别说很多课题组人员流失严重,技术断层现象普遍,导致不是课题进展缓慢,文章发不出去了或者被抢发,甚至几百万科研经费就打水漂。
在做知识整理和分享的这6年,类似的现象和求助我早日见怪不怪了!大多数情况下,我能做的真的很少,虽然大家求助的时候看起来都情深意切,但是我们双方都知道,我并不提供科研服务也不可能全身心投入到本来也不属于你的课题,作为咨询者的你,一份简陋的邮件很的可能上其实就是病急乱求医罢了。
如果你恰好有不确定的测序课题设计问题,恰好有不确定的数据分析流程的步骤软件参数阈值选择问题,或者其它觉得我可以帮到你的,而且你又恰好在这个时间段,在上海(11月23号周六下午3-6点,上海张江高科),可以过来参加我们的生物信息学的“义诊”。义诊规则
既然说了是“义诊”,那必须是免费的啦!不过,我们交流的形式是公开的,所以如果你的课题是高度机密,请留意明天的通知,这个“义诊”就不适合你啦!
时间肯定是不能变化的,我大老远飞去上海,总共就十几个小时时间,必须是你迁就我!
具体地址我还没有确定,根据参加人数来临时决定,肯定是在上海张江高科区域啦,毕竟我在那边工作生活了一年多, 还是蛮有感情的!
因为是第一次,所以我也不确定会是一种什么样的形式,你我将共同创造历史!
申请规则也非常简单,因为是“义诊”所以需要加上一点门槛,你需要邮件(jmzeng1314@163.com)写清楚你的问题,我会根据问题的价值,凭我自己的感觉来挑选朋友参加我们的义诊活动!如果你确实时间地点都不合适参加我们的义诊,也可以考虑根据下面的规则来邮件(jmzeng1314@163.com)提问,也有一定几率能获得答疑!
下面是一些实例
比如代码细节问题:
1、比对率低于多少这个样本可以剔除掉,因为在hisat2比对的时候有几个样本就77%,有几个是85%,其他基本在90%以上(绝大多数在95%以上),你这边有没有可供参考的文章 2、nohup重定向中,nohup ./start.sh & 默认输出到nohup.out文件 nohup ./start.sh >output 2>&1 & 指定输出到output文件 如果追加的话是nohup ./start.sh >>output 2>&1 & 但是2>&1是什么意思,因为在网上看的时候有些人又不加这个,是因为循环的问题吗 3、如果用trim_galore用同样的参数对样本【已经用相同参数处理过一次】再处理,结果是不是不会变化 4、同时运行相同的命令会出现问题吗:因为在运行的过程中一时没注意同时运行了如下命令(类似):samtools sort -@ 20 -o test.sort.bam test.bam 和 samtools sort -@ 30 -o test.sort.bam test.bam;最后结果是3g,然后我后来单独运行后的结果也是3g。我就默认是一致了,想问我的想法是对的吗,因为按理说应该是覆盖的关系,但是这种同时运行我就不清楚了。 5、sed -n '1,217p' bam.txt | while read id;do (nohup samtools sort -@ 30 -o ${id%.*}.sort.bam $id );done 在前台运行的程序ctrl + z暂停后然后用bg %1后台挂起运行,但只运行了一会,进程就消失了,这是为什么。比如流程选择问题:
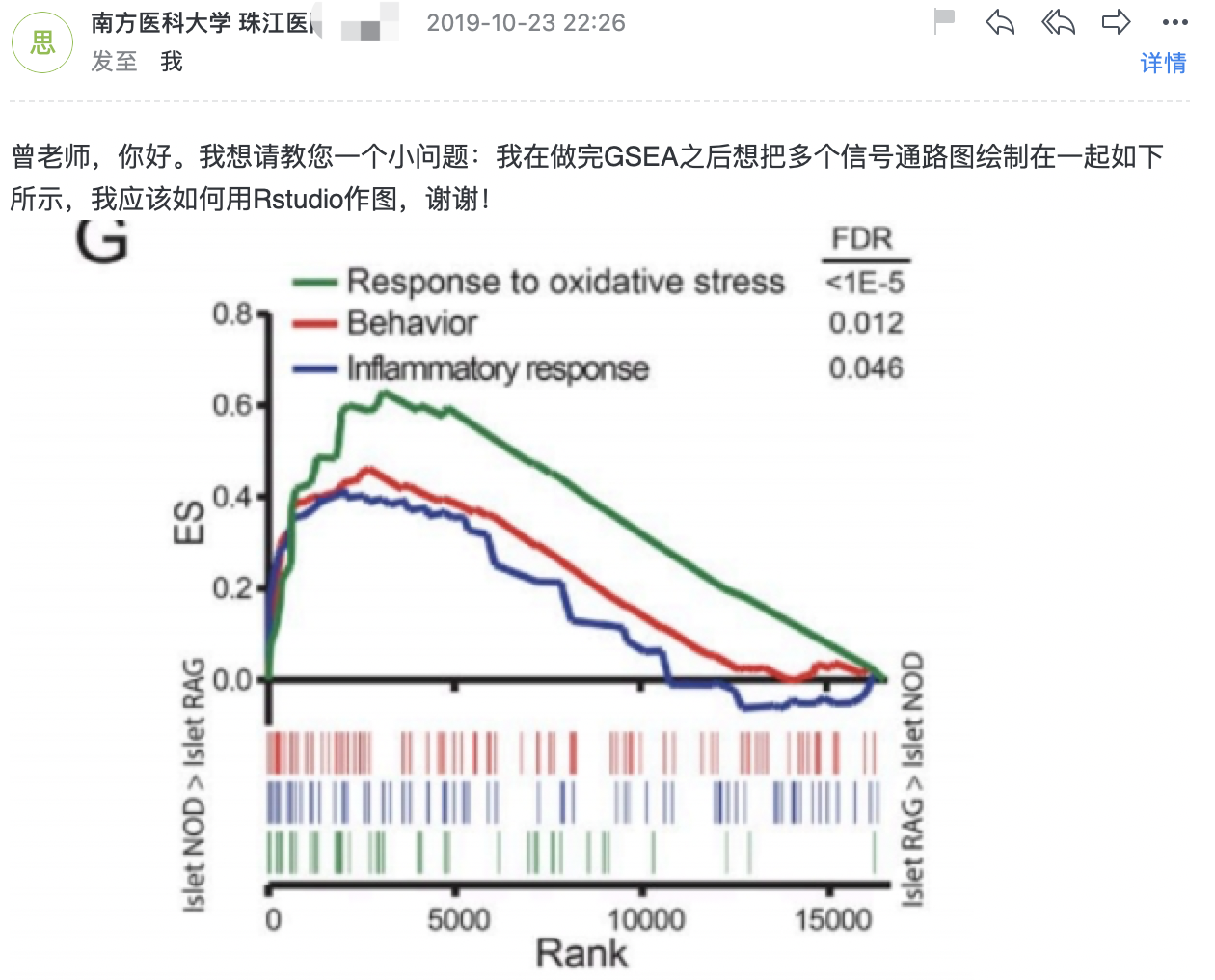
我现在在做六组rnaseq的特异基因分析(看每一组相对其他组的高表达),deseq2的统计原理是t-test,并不适用,我觉得应该用anova的统计方法。请问我该用什么方法呢? 现在我知道的是有一个k-means分组,还有deseq2老版本里面有一个anova函数但是新版本中已经删掉了,不知道这两种方法科学合理吗?或者有其它应该用的方法?或者是绘图问题,做完GSEA之后想把多个信号通路图绘制在一起:
比如网页工具问题:1.现在官方CIBERSORT网站,无法上传数据,请问有没有什么办法可以使用CIBERSORT 2.我们实验室有一组肿瘤的芯片表达谱,我想比较这组芯片和TCGA上的RNAseq得到的免疫浸润情况,请问是否可行,及对数据的处理方法。比如数据分析实战问题:
使用getGEO下载GSE129816数据时下载得到的矩阵文件,在执行exprSet=exprs(gset)这一步时得到的是一个空的矩阵。我看了一下文件大小就不对,并不是下载时出了问题,直接打开后里面也没有具体每个样本的表达矩阵。 然后我直接下载原始数据,他给出的原始数据是excel形式的,附件里有。使用平台是GPL13112,这个平台我没有找到对应的R包,而且原始文件给出的好像不是探针号,使用getGEO('GPL13112',destdir =".")后,Table(GPL13112)依然得到一个空的矩阵,我尝试了一下这个平台其他的一些测序数据,都是这样,没法进行下一步了,我想问下像这种数据集该如何分析。 我是北京协和医学院一名研二在读学生,看了您的教程后在尝试分析一些跟我课题相关的数据也可以是资源申请,我这里有几百个T的生物信息学资料,基本上你想要的,我这里都有,如果没有,我也可以找十万粉丝帮你收集!
同时招收志愿者
因为我也没有组织过义诊,而且已经好些年没有回上海了,不是很熟悉那边的情况,如果你想全程参与,深度参与我们的义诊活动,欢迎报名参加志愿者!同样的是需要邮件(jmzeng1314@163.com)申请,志愿者至少可以获得无限生物信息学资料吧,还有优先提问几乎一个!