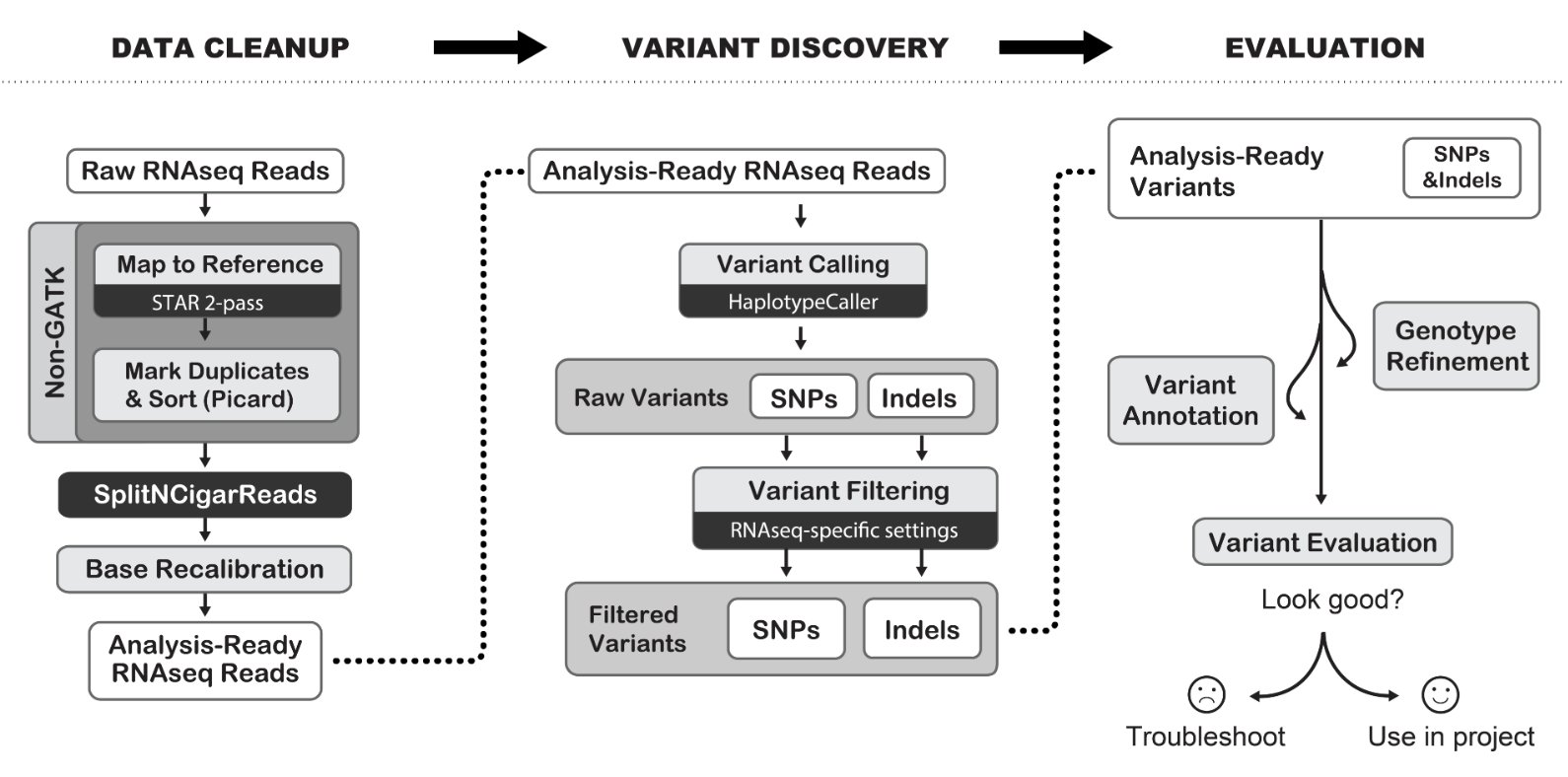
RNA-seq标准分析,我们已经讲解的太多了,表达矩阵到差异分析等下游生物学注释都没有啥新颖之处,融合基因和可变剪切算是出彩的地方,如果加上GATK找变异流程就更棒了,反正都使用了star软件进行序列比对拿到bam文件了。
如果你简单谷歌搜索关键词:gatk best practices pipeline rna-seq 会搜索到大量过期的教程:
- 2014年5月6日 - Best Practices workflow for RNAseq https://gist.github.com/PoisonAlien/c6c03539cf4b1ac41cf1
- 2016年10月27日 - https://www.biostars.org/p/219281/
- 2016年10月17日 - https://gatkforums.broadinstitute.org/gatk/discussion/3892/the-gatk-best-practices-for-variant-calling-on-rnaseq-in-full-detail
- 2017年3月17日 - 2017 Mar 17. doi: 10.12688/wellcomeopenres.10501.2
因为软件和数据库都是在持续更新,所以我们必须得紧跟潮流。
我们需要看最新的:Best Practices Workflows | Created 2018-01-09 | Last updated 2019-07-11 ,链接是 https://software.broadinstitute.org/gatk/best-practices/workflow?id=11164
回顾一下star的2-pass比对代码
我在介绍star-fusion:最好用的融合基因查找工具终于正式发表了 的时候,附带了比对代码,核心代码是:
start=$(date +%s.%N)
echo star start `date`
$bin_star --runThreadN 4 --genomeDir $star_index \
--twopassMode Basic --outReadsUnmapped None --chimSegmentMin 12 \
--alignIntronMax 100000 --chimSegmentReadGapMax parameter 3 \
--alignSJstitchMismatchNmax 5 -1 5 5 \
--readFilesCommand zcat --readFilesIn $fq1 $fq2 --outFileNamePrefix ${sample}_
mv ${sample}_Aligned.out.sam $sample.sam
$bin_samtools sort -o $sample.bam $sample.sam
$bin_samtools index $sample.bam
$bin_samtools flagstat $sample.bam > $sample.flagstat
# 这里需要判断上一个步骤是否成功,判断命令状态
touch ok.star.$sample.status
rm $sample.sam
echo star end `date`
dur=$(echo "$(date +%s.%N) - $start" | bc)
printf "Execution time for star : %.6f seconds" $dur
实际上就是一行命令在运行比对过程,但是呢,参数太多了,调起来很麻烦,通常如果不理解的话就不建议修改参数。如果你确实需要做star-fusion 有一个链接需要注意:https://github.com/STAR-Fusion/STAR-Fusion/issues/104 我们这里仅仅是需要star比对后的bam文件来走GATK找变异流程。
参考基因组都使用star-fusion的31G数据库文件里面的:
~/biosoft/starFusion/db/GRCh38_gencode_v31_CTAT_lib_Oct012019.plug-n-play/ctat_genome_lib_build_dir
值得注意的是,因为下载star-fusion的31G数据库文件解压后只有 ref_genome.fa ,并没有 ref_genome.dict,需要自己构建。
Picard CreateSequenceDictionary creates .dict file and samtools faidx creates a .fai file. Both are needed for GATK. 自行搜索如何构建哈。
下载最新版GATK
官网:https://software.broadinstitute.org/gatk/ 目前最新版是:4.1.4.0 我后面的教程都是基于此:
地址是:https://github.com/broadinstitute/gatk/releases/download/4.1.4.0/gatk-4.1.4.0.zip
mkdir -p ~/biosoft
cd ~/biosoft
mkdir gatk
wget https://github.com/broadinstitute/gatk/releases/download/4.1.4.0/gatk-4.1.4.0.zip
unzip gatk-4.1.4.0.zip
主要流程
首先去除PCR重复
conda activate rna
# 我们对bam文件取部分作为测试数据
samtools view -h ../star/SRR2016932.bam|head -100000 > tmp.sam
samtools sort -O bam -@ 4 tmp.sam -o tmp.bam
samtools index tmp.bam
conda install -y -c bioconda sambamba
sample=tmp
sambamba markdup --overflow-list-size 600000 --tmpdir='./' -r $sample.bam ${sample}_rmd.bam
然后SplitNCigarReads
这个时候需要参考基因组文件,以及配套的dict好fai文件。
gatk='/home/jmzeng/biosoft/gatk/gatk-4.1.4.0/gatk'
GENOME='/home/jmzeng/biosoft/starFusion/db/GRCh38_gencode_v31_CTAT_lib_Oct012019.plug-n-play/ctat_genome_lib_build_dir/ref_genome.fa'
$gatk CreateSequenceDictionary -R $GENOME # 最新版gatk,这个步骤非常快
$gatk SplitNCigarReads -R $GENOME \
-I ${sample}_rmd.bam \
-O ${sample}_rmd_split.bam
这样得到的bam文件,才跟DNA流程的类似,可以走gatk后续流程了。
接着走Base Quality Recalibration
gatk='/home/jmzeng/biosoft/gatk/gatk-4.1.4.0/gatk'
DBSNP=/home/jmzeng/biosoft/GATK/resources/bundle/hg38/dbsnp_146.hg38.vcf.gz
kgSNP=/home/jmzeng/biosoft/GATK/resources/bundle/hg38/1000G_phase1.snps.high_confidence.hg38.vcf.gz
kgINDEL=/home/jmzeng/biosoft/GATK/resources/bundle/hg38/Mills_and_1000G_gold_standard.indels.hg38.vcf.gz
GENOME='/home/jmzeng/biosoft/starFusion/db/GRCh38_gencode_v31_CTAT_lib_Oct012019.plug-n-play/ctat_genome_lib_build_dir/ref_genome.fa'
# 因为我的star比对得到的bam文件里面没有 Read groups
# 参考:https://gatkforums.broadinstitute.org/gatk/discussion/6472/read-groups
$gatk AddOrReplaceReadGroups -I ${sample}_rmd_split.bam \
-O ${sample}_rmd_split_add.bam -LB $sample -PL ILLUMINA -PU $sample -SM $sample
$gatk --java-options "-Xmx20G -Djava.io.tmpdir=./" BaseRecalibrator \
-I ${sample}_rmd_split_add.bam \
-R $GENOME \
-O ${sample}_recal.table --known-sites $kgSNP --known-sites $kgINDEL
$gatk --java-options "-Xmx20G -Djava.io.tmpdir=./" ApplyBQSR \
-I ${sample}_rmd_split_add.bam -R $GENOME --output ${sample}_recal.bam -bqsr ${sample}_recal.table
最后Variant Calling
因为最后所有的样本需要合并比较,所以这里使用gvcf模式的Variant Calling
bed=/home/jmzeng/annotation/CCDS/human/exon_probe.GRCh38.gene.150bp.bed
# ftp://ftp.ncbi.nlm.nih.gov/snp/organisms/human_9606/VCF/All_20180418.vcf.gz
bam=${sample}_recal.bam
$gatk --java-options "-Xmx20G -Djava.io.tmpdir=./" HaplotypeCaller \
-ERC GVCF -L $bed -R $GENOME -I $bam --dbsnp $DBSNP -O ${sample}_gatk.gvcf
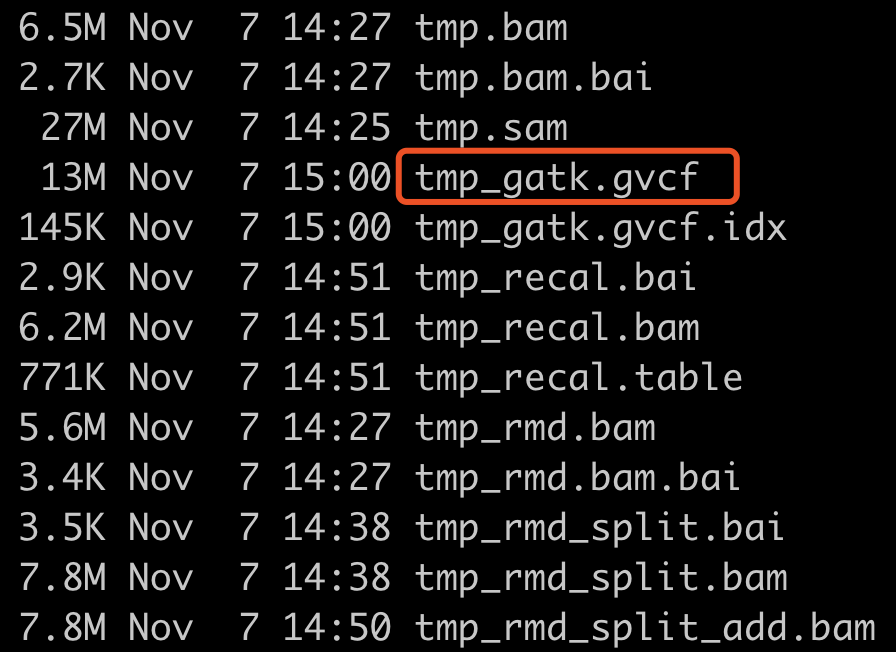
一个样本的star比对后的bam文件,走RNA-seq数据的GATK找变异流程得到的全部文件如下:
如果是普通的vcf文件,是需要过滤一下的,官网是这样说的:We recommend specific hard filters, since VQSR and CNNScoreVariants require truth data for training that we don’t yet have for RNA.
因为我这里走的是gvcf模式,所以暂时不需要这个过滤步骤。
串起来成为shell脚本流程
只需要把我们的全部star的bam文件全路径保存为bam_path.txt 文件,就可以使用下面的脚本提交任务批量运行我们的gvcf流程啦,当然也可以控制并行的样本数量,具体需要理解shell脚本的语法了。
cat bam_path.txt |while read id
do
file=$(basename $id )
sample=${file%%.*}
if((i%$number1==$number2))
then
if [ ! -f ok.gvcf.$sample.status ]; then
time sambamba markdup --overflow-list-size 600000 --tmpdir='./' -r $id ${sample}_rmd.bam
time $gatk SplitNCigarReads -R $GENOME \
-I ${sample}_rmd.bam \
-O ${sample}_rmd_split.bam
time $gatk AddOrReplaceReadGroups -I ${sample}_rmd_split.bam \
-O ${sample}_rmd_split_add.bam -LB $sample -PL ILLUMINA -PU $sample -SM $sample
time $gatk BaseRecalibrator \
-I ${sample}_rmd_split_add.bam \
-R $GENOME \
-O ${sample}_recal.table --known-sites $kgSNP --known-sites $kgINDEL
time $gatk ApplyBQSR \
-I ${sample}_rmd_split_add.bam -R $GENOME --output ${sample}_recal.bam -bqsr ${sample}_recal.table
time $gatk HaplotypeCaller \
-ERC GVCF -L $bed -R $GENOME -I ${sample}_recal.bam --dbsnp $DBSNP -O ${sample}_gatk.gvcf
if [ $? -eq 0 ]; then
echo "succeed" $sample
touch ok.gvcf.$sample.status
rm ${sample}_rmd.bam ${sample}_rmd_split.bam ${sample}_rmd_split_add.bam
rm ${sample}_rmd.bam.bai ${sample}_rmd_split.bam.bai
else
echo "failed" $sample
fi # check gvcf status
fi ## check whether we need to process current sampel
fi # check the batch
i=$((i+1))
done
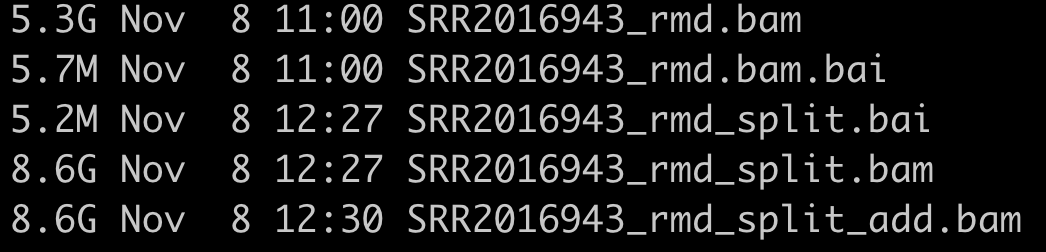
一个真正的样本测序数据走这个针对RNA-seq数据的GATK找变异流程会输出的文件非常多,而且耗时很夸张的!
下面的5个文件是需要删除的:
得到vcf文件,目前不显示了。
大家注意一下java问题
仔细看我分步骤调试代码的时候,有时候加上了 " --java-options -Xmx20G -Djava.io.tmpdir=./" 的参数,但是合并起来成为脚本的时候,又删除它了,具体细节看: