RNA-seq数据毫无疑问是目前NGS领域被使用最频繁的了,但是大部分科研人员对它的理解,还停留在表达量层面,尤其是基于基因的表达量,无非就是分组,然后走差异分析这样的统计学检验,绘制火山图和差异基因热图,上下调的通路。
先不说大家对RNA-seq数据的标准分析是否一定是对的,这样的简陋的分析其实是对数据的暴殄天物!
首先可以分析差异转录本,可变剪切
看到一篇2019年5月发表在Molecular Neurodegeneration杂志的文章:TREM2 brain transcript-specific studies in AD and TREM2 mutation carriers 把普通的RNA-seq数据根据自己的生物学背景挖掘了一下。背景知识需要去搜索了解Triggering Receptor Expressed in Myeloid cells 2 (TREM2)这个基因,以及它的3个转录本。
都是European-Americans,测序数据是:
- AD cases with TREM2 variants (n = 33)
- AD cases (n = 195)
- healthy controls (n = 118)
来源于3个不同的机构: - Washington University in St. Louis Knight-ADRC Brain Bank (51 participants)
- MSBB-BM36, (132 participants)
- MCBB (162 participants)
每个样本平均测序数据量是 134.9 million ,是2 × 101bp的测序策略。
其中2个机构的数据是已有的,数据下载方式: - Mayo Clinic Brain Bank RNA-seq data was downloaded from the AMP-AD portal (synapse ID = 5,550,404; accessed January 2017).
- Mount Sinai Brain Bank RNA-seq data was downloaded from the AMP-AD portal (synapse ID = https://www.synapse.org/#!Synapse:syn3157743; accessed January 2017).
转录组数据分析流程,主要是软件选择,参考基因组版本: - FastQC [49] was used to assess sequencing quality.
- The RNA-seq was aligned to the human GRCh37 primary assembly using STAR (ver 2.5.2b).
- Read alignments were further evaluated by using PICARD CollectRnaSeqMetrics (ver 2.8.2) to examine read distribution across the genome.
- We employed Kallisto (v0.42.5) and tximport to determine the read count for each transcript and quantified transcript abundance as transcripts per kilobase per million reads mapped (TPM), using gene annotation of Homo sapiens reference genome (GENCODE GRCh37) for each participant from Knight-ADRC, MCB and MSBB-BM36 independently, with the following parameters: -t 10 -b 100.
- Then we summed the read counts and TPM of all alternative splicing transcripts of a gene to obtain gene expression levels. Due to the positive skewness of TPM values, we calculate their logarithm10 (log10TPM) for further analysis.
关于转录本的差异分析,我们分享过salmon+DRIMseq流程,在前些天的推文里面,见:每月一生信流程之rnaseqDTU(差异转录本)
在文章导论大量介绍了TREM2)这个基因,以及它的3个转录本。同时看了3个队列的这个基因的3个转录本的表达量情况。We were able to detect and quantify the levels of three TREM2 transcripts ENST00000373113, ENST00000373122 and ENST00000338469 using RNA-seq data from AD and control brains from three different, independent studies.
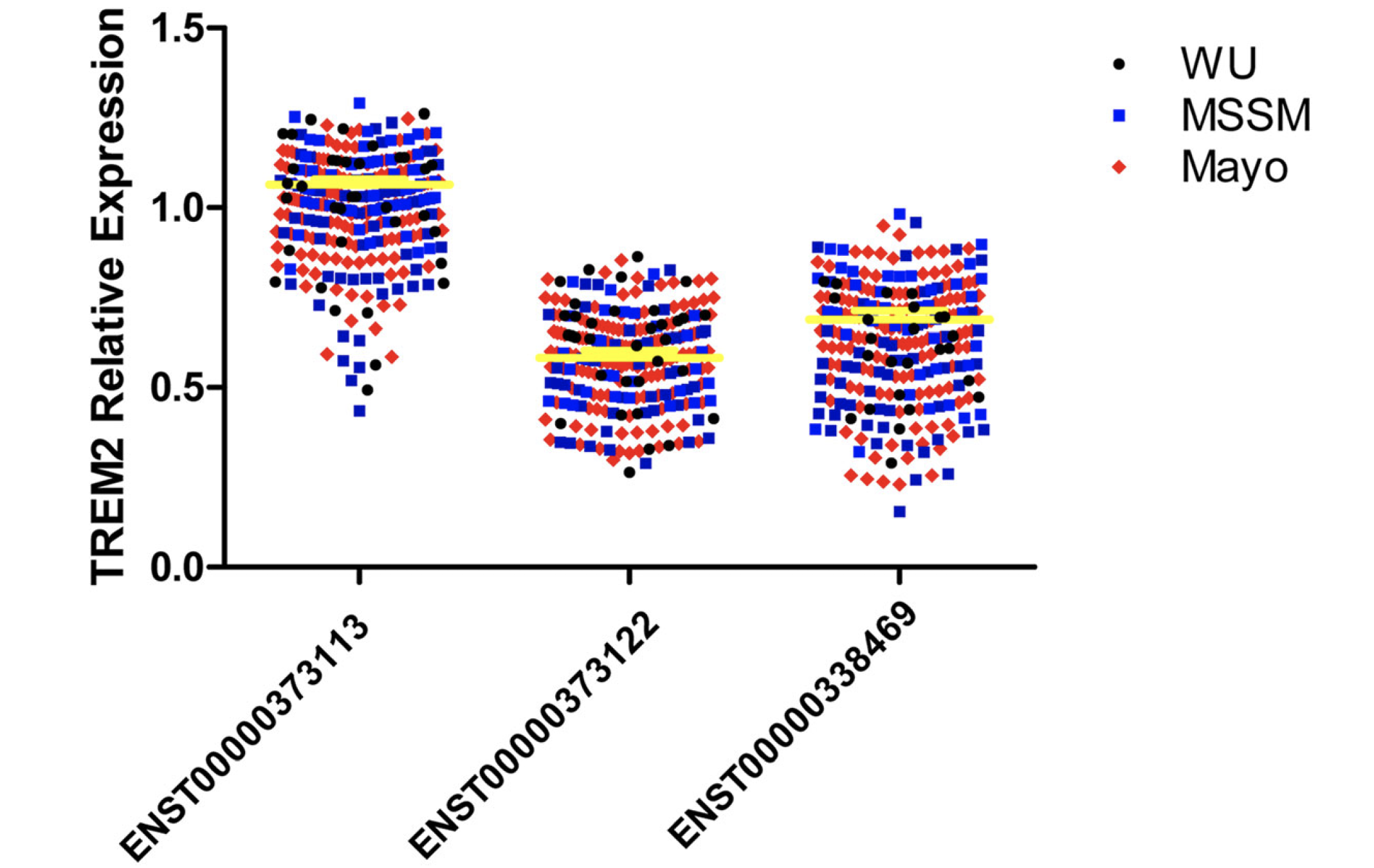
不过这样的分析仍然是片面的,因为作者仅仅是关心自己生物学背景的基因,下面的全局比较的总结表格其实是不可或缺的。
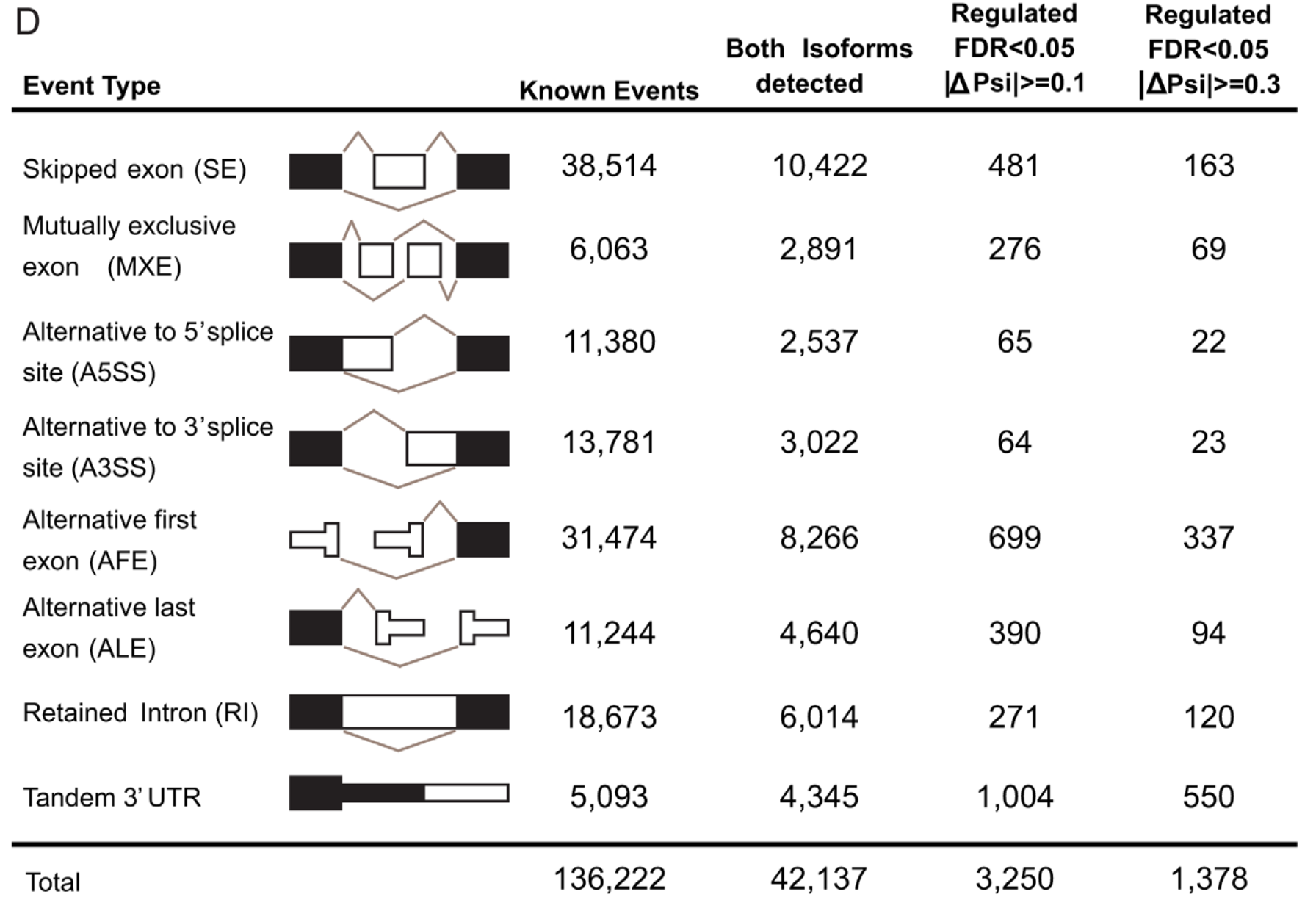
然后可以分析融合基因
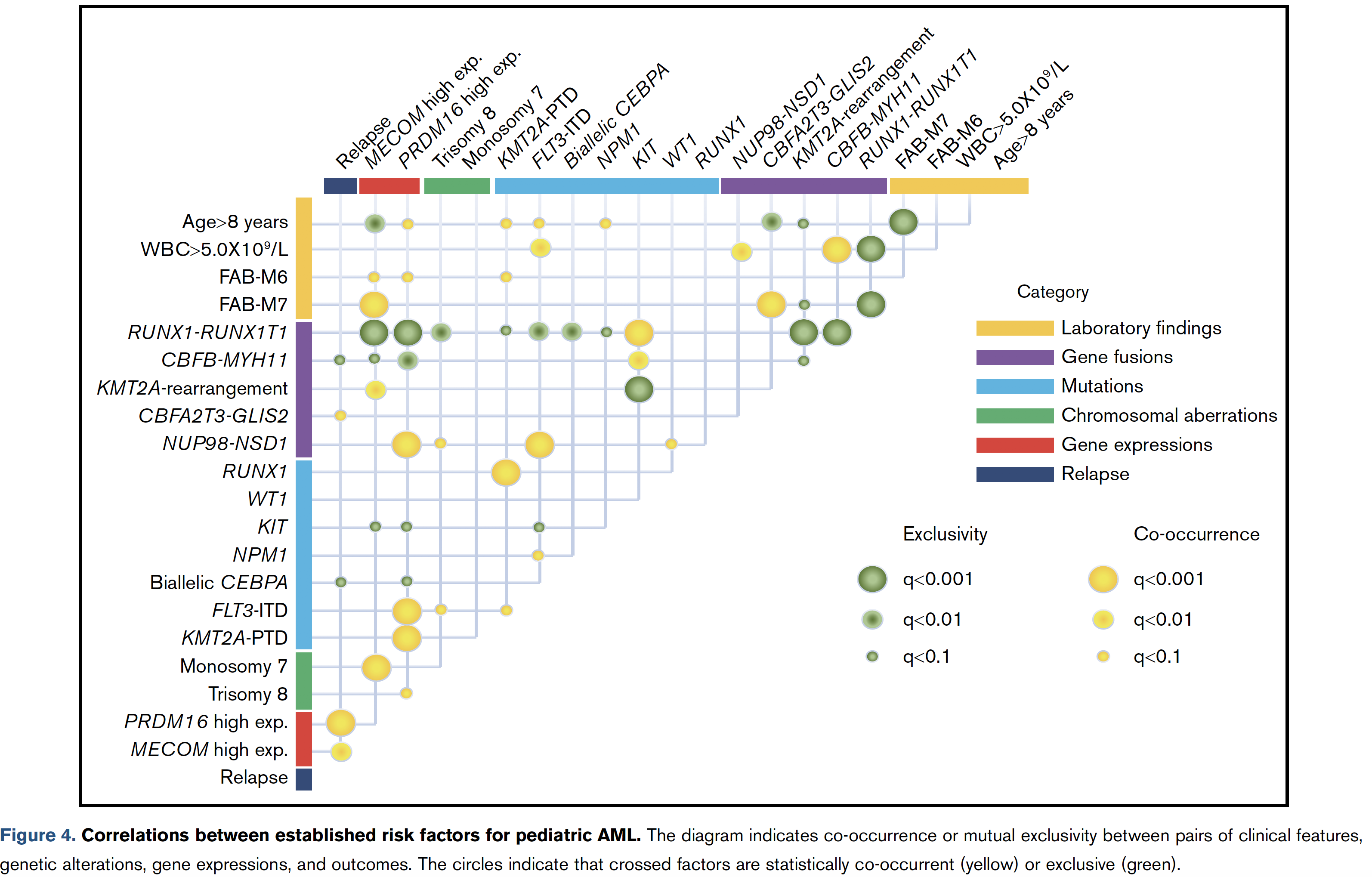
看到[article] (2019) Transcriptome analysis offers a comprehensive illustration of the genetic background of pediatric acute myeloid leukemia. Blood Adv 文章就是日本研究团队的 [RNA-seq] in 139 of the 369 patients with de novo pediatric AML ,这样文章落脚点就是基因融合事件,54 in-frame gene fusions and 1 RUNX1 out-of-frame fusion in 53 of 139 patients.
在大的病人队列里面,提供实验验证了 258 gene fusions in 369 patients (70%) 。
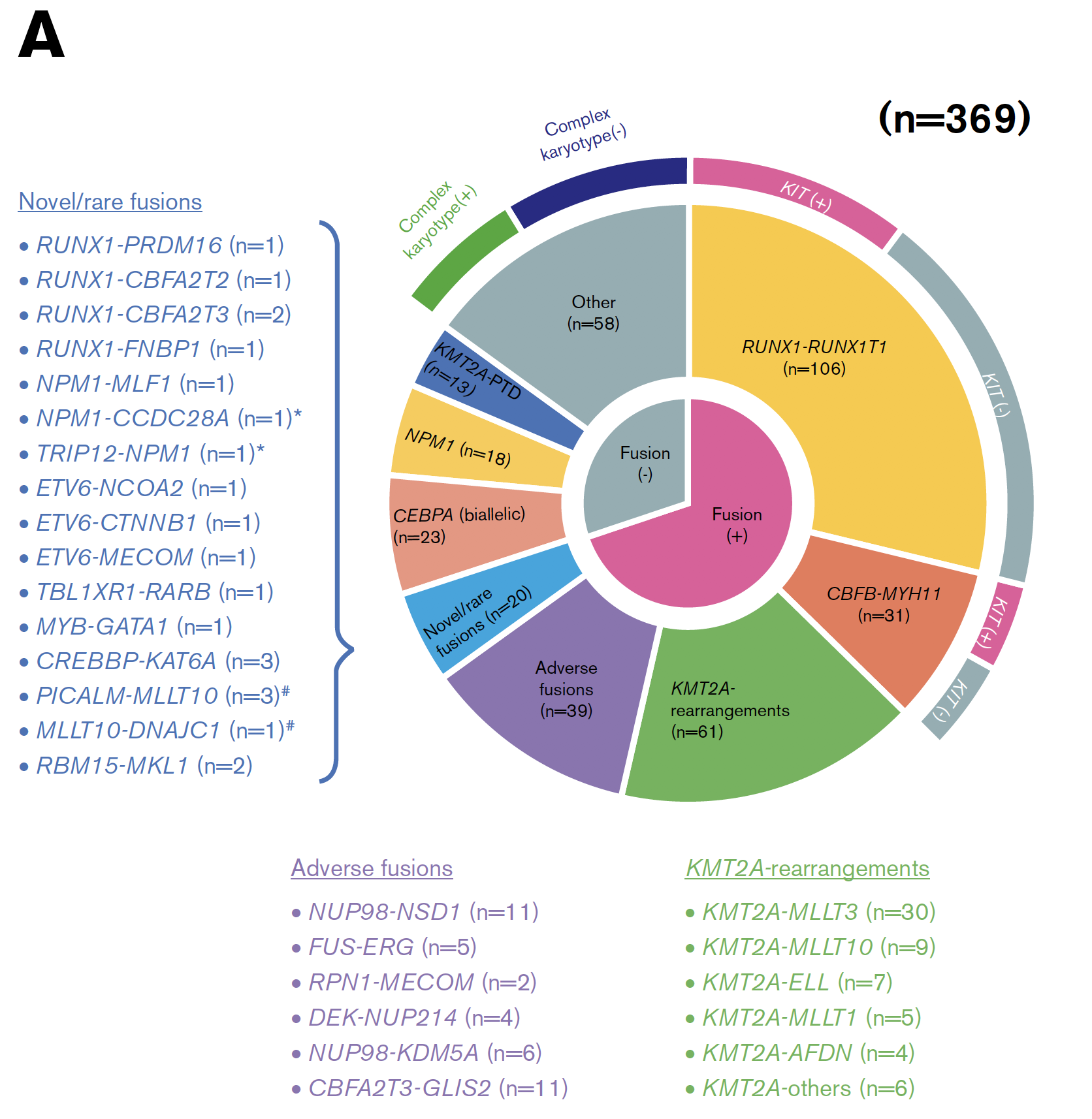
因为有RNA-seq数据的只有139个病人,所以 突变全景图如下:
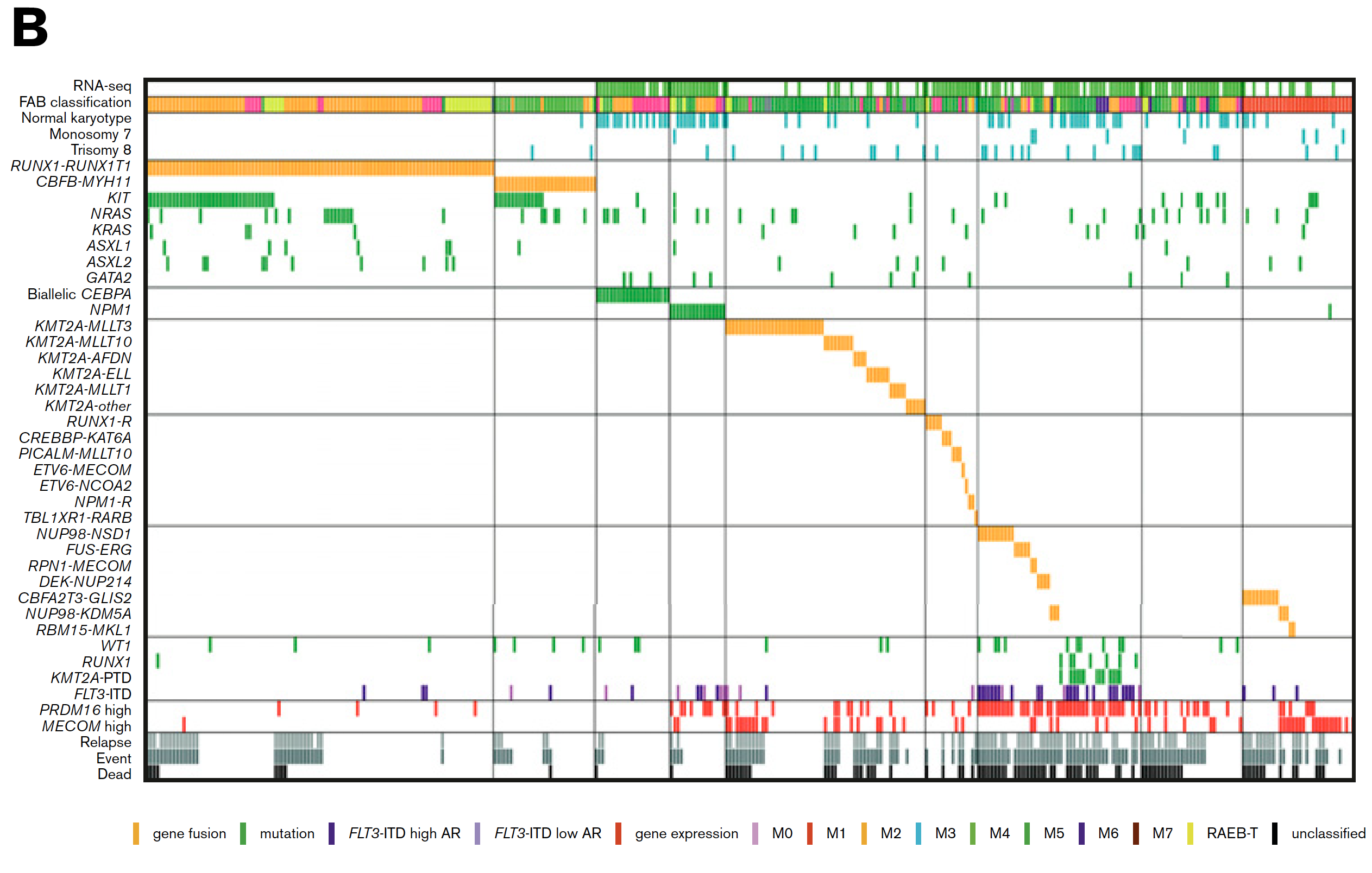
甚至找到的基因融合事件,可以当做是病人的一种表型信息进行分析:
关于可变剪切和融合基因的RNA-seq数据分析教程
因为做目录确实很浪费时间,差不多就下面这些,大家先学习吧:
- 100篇泛癌研究文献解读之可变剪切事件大起底
- rMATS这款差异可变剪切分析软件的使用体验
- 用LeafCutter探索转录组数据的可变剪切
- 用Expedition来分析单细胞转录组数据的可变剪切
- 使用SGSeq探索可变剪切
- 用DEXSeq分析可变剪切,外显子差异表达
- miRNA、LncRNA、CircRNA靠谱小结
- 超2万样本的RNA-seq数据重新统一处理(TCGA+GTEx+ TARGET)
- 玩转RNA-seq数据也可以不需要linux ?
- 高表达的PVT1(lncRNA)能够独立且有效地预测葡萄膜黑色素瘤生存情况
- RNA-seq技术已经常规化,你还好意思不掌握吗?
最后友情宣传生信技能树
- 生物信息学“义诊”
- 生物信息学”拍卖会”
- 全国巡讲:R基础,Linux基础和RNA-seq实战演练 : 预告:12月28-30长沙站
- 广州珠江新城GEO数据挖掘滚动开班
- DNA及RNA甲基化数据分析与课题设计