本来有statquest珠玉在前,我实在是提不起笔和勇气写统计学专题,但是最近直播单细胞转录组数据分析发现这系列知识点实在是太重要,而我的习惯是,讲不清楚的知识点不认为自己掌握了,所以还是尝试着介绍一波。
统计基础
统计学其实可以分为两大类:
- 一:描述性统计,充分了解你的数据,分析数据的
集中趋势和离散趋势等统计学指标并且可视化 - 二:推断统计学,根据
样本数据去推断总体数量特征的方法。它是在对样本数据进行描述的基础上,对统计总体的未知数量特征做出以概率形式表述的推断。
数据总体来说可以分为以下三种类型:
- 一:分类数据,又名定性数据或者品质数据。
- 二:顺序数据,它其实是是分类数据的一种。
- 三:数值型数据,又名定量数据,这个才是重点,又可以分成离散型和连续型。
定量数据的集中趋势指标主要是:众数、分位数和平均数
定量数据的离散趋势指标主要是:极差,方差和标准差,标准分数,相对离散系数(变异系数),偏态系数与峰态系数
载入表达矩阵及分组信息
首先看看 airway 数据集
options(stringsAsFactors = F)
library(airway)
data(airway)
# 这里需要自行学习bioconductor里面的RangedSummarizedExperiment对象
airway
## class: RangedSummarizedExperiment
## dim: 64102 8
## metadata(1): ''
## assays(1): counts
## rownames(64102): ENSG00000000003 ENSG00000000005 ... LRG_98 LRG_99
## rowData names(0):
## colnames(8): SRR1039508 SRR1039509 ... SRR1039520 SRR1039521
## colData names(9): SampleName cell ... Sample BioSample
RNAseq_expr=assay(airway)
colnames(RNAseq_expr)
## [1] "SRR1039508" "SRR1039509" "SRR1039512" "SRR1039513" "SRR1039516"
## [6] "SRR1039517" "SRR1039520" "SRR1039521"
RNAseq_expr[1:4,1:4]
## SRR1039508 SRR1039509 SRR1039512 SRR1039513
## ENSG00000000003 679 448 873 408
## ENSG00000000005 0 0 0 0
## ENSG00000000419 467 515 621 365
## ENSG00000000457 260 211 263 164
# RNAseq_expr 是一个数值型矩阵,属于连续性变量,可以探索众数、分位数和平均数 ,极差,方差和标准差等统计学指标
RNAseq_gl=colData(airway)[,3]
table(RNAseq_gl)
## RNAseq_gl
## trt untrt
## 4 4
# RNAseq_gl 是一个分类变量
可以看到总共是8个样本的RNA-seq数据的counts矩阵,这8个样本分成2组,每组是4个样本, 分别是 trt 和 untrt 组。
然后看看 ALL 数据集
library(ALL)
data(ALL)
ALL
## ExpressionSet (storageMode: lockedEnvironment)
## assayData: 12625 features, 128 samples
## element names: exprs
## protocolData: none
## phenoData
## sampleNames: 01005 01010 ... LAL4 (128 total)
## varLabels: cod diagnosis ... date last seen (21 total)
## varMetadata: labelDescription
## featureData: none
## experimentData: use 'experimentData(object)'
## pubMedIds: 14684422 16243790
## Annotation: hgu95av2
# 这里需要自行学习bioconductor里面的 ExpressionSet 对象
array_expr=exprs(ALL)
# array_expr 是一个数值型矩阵,属于连续性变量,可以探索众数、分位数和平均数 ,极差,方差和标准差等统计学指标
array_pd=pData(ALL)
# array_pd 在这里是一个非常复杂的数据对象,每一列的数据类型不一样,可以进行多种多样的探索。
DT::datatable(array_pd,
extensions = 'FixedColumns',
options = list(
scrollX = TRUE,
fixedColumns = TRUE
))
可以看到临床信息很丰富:
fivenum函数看每个样本的五个分位数统计量
library(airway)
RNAseq_expr=assay(airway)
apply(RNAseq_expr,2,fivenum)
## SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516 SRR1039517
## [1,] 0 0 0 0 0 0
## [2,] 0 0 0 0 0 0
## [3,] 0 0 0 0 0 0
## [4,] 10 8 12 6 11 12
## [5,] 297906 255662 513766 273878 397791 401539
## SRR1039520 SRR1039521
## [1,] 0 0
## [2,] 0 0
## [3,] 0 0
## [4,] 9 8
## [5,] 378834 372489
# 可以看到每个样本都有大量的基因表达量是0,所以我们应该是需要对基因进行一定程度的过滤。
apply(RNAseq_expr,2, function(x) sum(x>1))
## SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516 SRR1039517
## 21133 20844 22007 19782 21974 22313
## SRR1039520 SRR1039521
## 21173 20740
# 平均每个样本被检测到的基因数量是2万左右。
dim(RNAseq_expr)
## [1] 64102 8
RNAseq_expr=RNAseq_expr[apply(RNAseq_expr,1, function(x) sum(x>1) > 5),]
dim(RNAseq_expr)
## [1] 19481 8
apply(RNAseq_expr,2,fivenum)
## SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516 SRR1039517
## [1,] 0 0 0 0 0 0
## [2,] 18 15 22 12 20 23
## [3,] 181 151 209 113 204 237
## [4,] 737 665 843 496 850 1036
## [5,] 297906 255662 513766 273878 397791 401539
## SRR1039520 SRR1039521
## [1,] 0 0
## [2,] 17 16
## [3,] 163 161
## [4,] 659 709
## [5,] 378834 372489
boxplot看表达量分布整体情况
箱线图(Boxplot)也称箱须图(Box-whisker Plot)。
是利用数据中的五个统计量:最小值、第一四分位数、中位数、第三四分位数与最大值来描述数据的一种方法,它也可以粗略地看出数据是否具有有对称性,分布的分散程度等信息,特别可以用于对几个样本的比较。
apply(RNAseq_expr,2,fivenum)
## SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516 SRR1039517
## [1,] 0 0 0 0 0 0
## [2,] 18 15 22 12 20 23
## [3,] 181 151 209 113 204 237
## [4,] 737 665 843 496 850 1036
## [5,] 297906 255662 513766 273878 397791 401539
## SRR1039520 SRR1039521
## [1,] 0 0
## [2,] 17 16
## [3,] 163 161
## [4,] 659 709
## [5,] 378834 372489
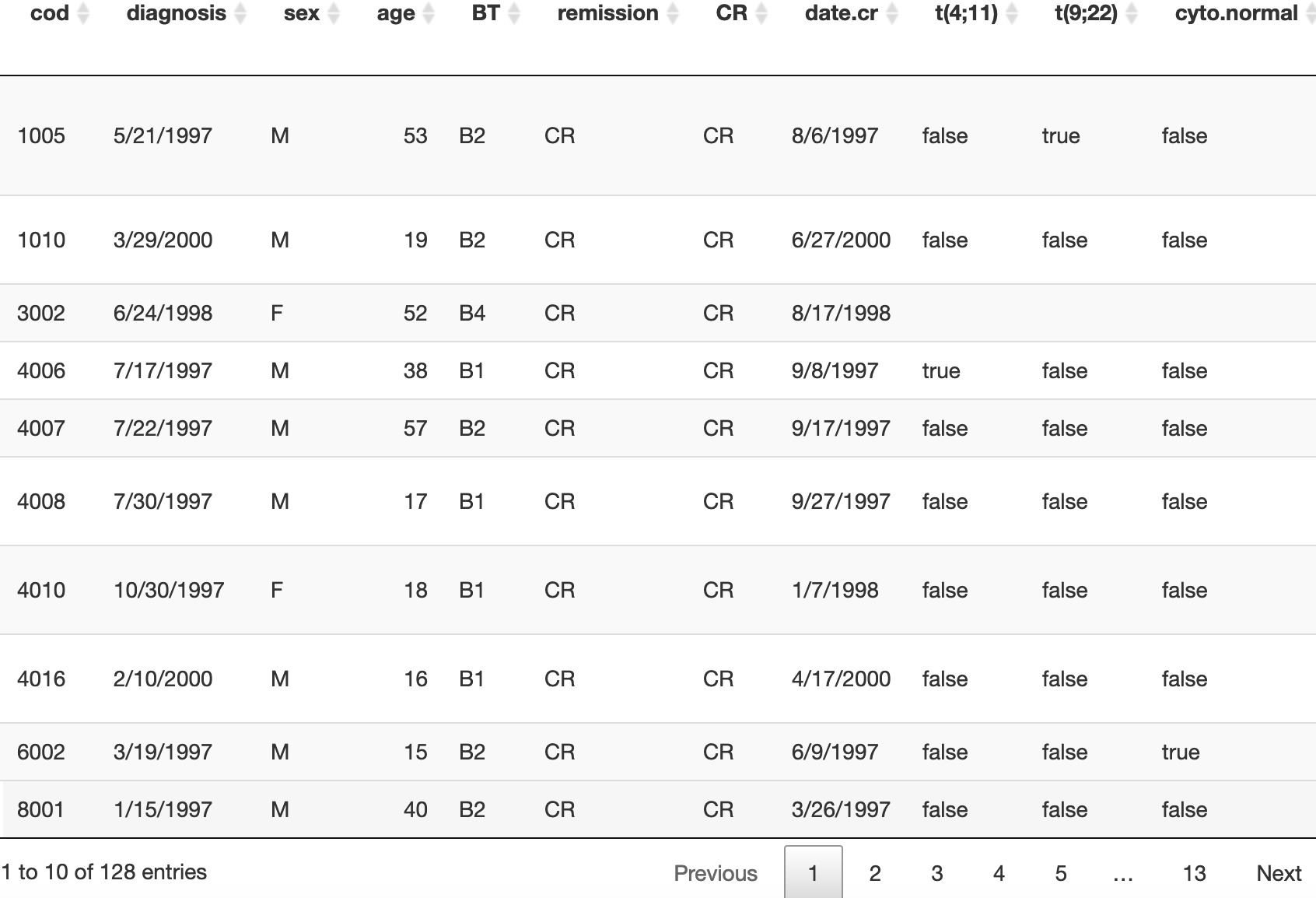
boxplot(RNAseq_expr,las=2)
可以看到表达量是原始count值,高表达量基因太大。
# 因为基因表达量现在是counts矩阵,所以超高表达量的基因会影响坐标轴,导致箱线图的箱子几乎看不清。
RNAseq_expr=log2(RNAseq_expr+1)
apply(RNAseq_expr,2,fivenum)
## SRR1039508 SRR1039509 SRR1039512 SRR1039513 SRR1039516 SRR1039517
## [1,] 0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
## [2,] 4.247928 4.000000 4.523562 3.700440 4.392317 4.584963
## [3,] 7.507795 7.247928 7.714246 6.832890 7.679480 7.894818
## [4,] 9.527477 9.379378 9.721099 8.957102 9.733015 10.018200
## [5,] 18.184502 17.963884 18.970755 18.063179 18.601655 18.615184
## SRR1039520 SRR1039521
## [1,] 0.000000 0.000000
## [2,] 4.169925 4.087463
## [3,] 7.357552 7.339850
## [4,] 9.366322 9.471675
## [5,] 18.531210 18.506842
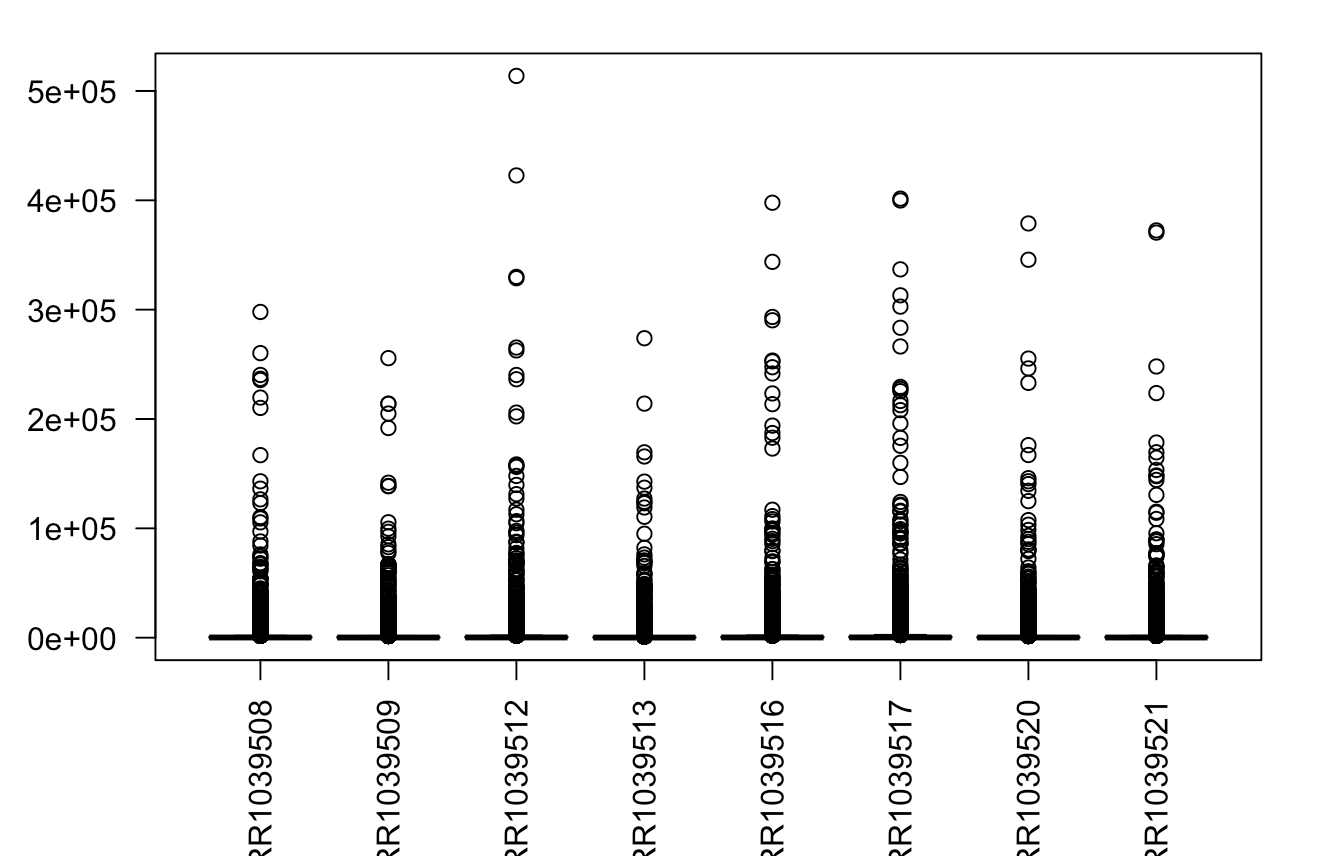
boxplot(RNAseq_expr,las=2)
简单的log后就看能看清楚每个样本的基因的表达量分布情况。
相关性
上面介绍的airway 数据集的表达矩阵 是RNA-seq技术得到的counts矩阵
options(stringsAsFactors = F)
library(airway)
data(airway)
exprSet=assay(airway)
colnames(exprSet)
## [1] "SRR1039508" "SRR1039509" "SRR1039512" "SRR1039513" "SRR1039516"
## [6] "SRR1039517" "SRR1039520" "SRR1039521"
group_list=colData(airway)[,3]
table(group_list)
## group_list
## trt untrt
## 4 4
tmp=data.frame(g=group_list)
rownames(tmp)=colnames(exprSet)
如果简单的使用全部基因来看样本间相关系数的分布情况
M=cor(exprSet)
# 可以看到这里的相关系数并不完全符合样本分组的
pheatmap::pheatmap(M,annotation_col = tmp)

有个小问题,就是相关系数实在是太高,而且它们的相关系数也没有根据分组来分布,不符合实际情况。
所以需要过滤基因再看相关性系数
exprSet=exprSet[apply(exprSet,1, function(x) sum(x>1) > 5),]
# 可以看到过滤了60%的低表达量基因后,相关系数显著下降
boxplot(cor(exprSet),las=2)
exprSet=log(edgeR::cpm(exprSet)+1)
# 但是归一化后相关系数又提高了。
boxplot(cor(exprSet),las=2)
接着取表达量变化大基因继续看样本的相关性, 这里我们采用 mad 指标来衡量基因的表达量的稳定情况。
sortGs=names(sort(apply(exprSet, 1,mad),decreasing = T))
boxplot(cor(exprSet[sortGs[1:100],]),las=2)
通过测试不同的基因数量的比较,最后确定取top500基因
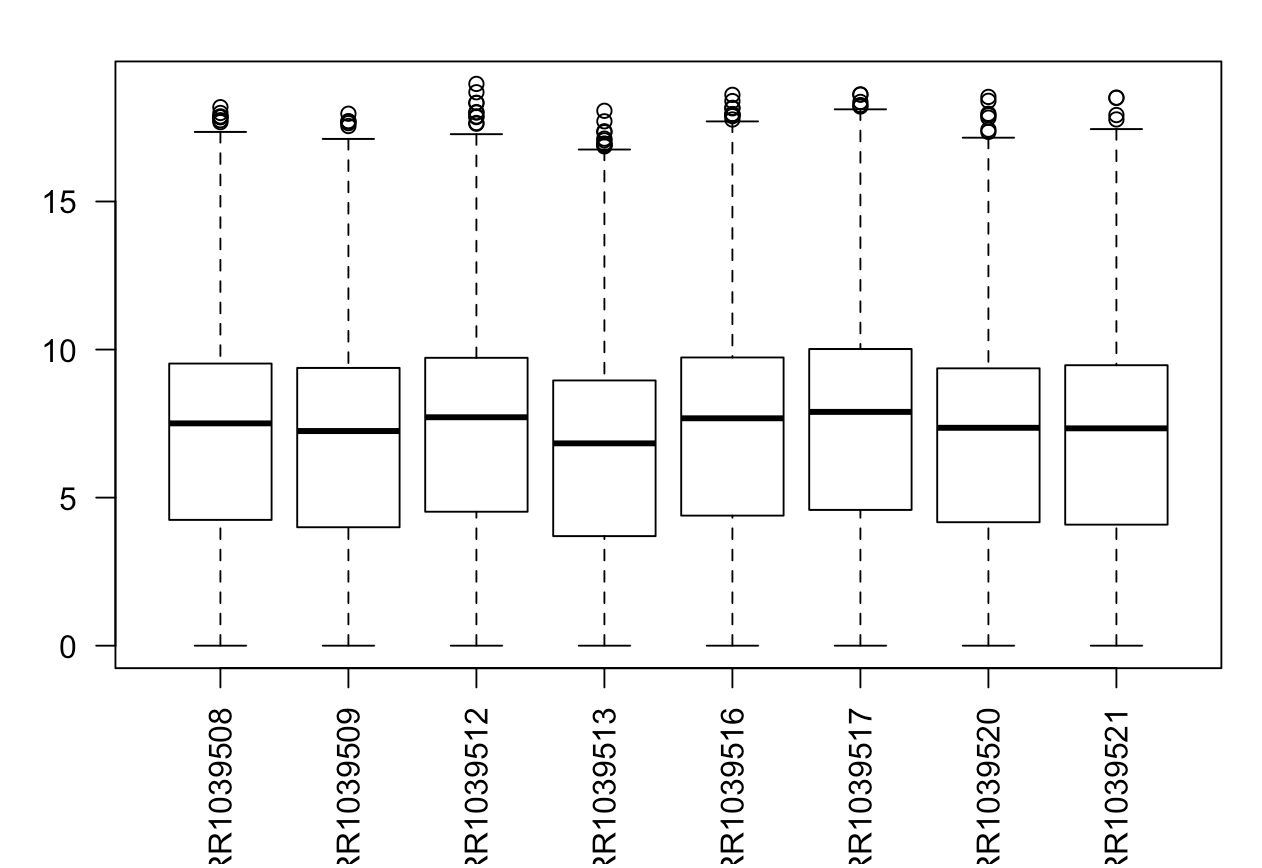
M=cor(exprSet[sortGs[1:500],])
pheatmap::pheatmap(M,annotation_col = tmp)
现在得到的相关性系数就符合样本的分组了。
quantile归一化的影响并不影响样本的相关性。
library(preprocessCore)
x=exprSet[sortGs[1:500],]
M1=cor(normalize.quantiles(x,copy=TRUE))
pheatmap::pheatmap(cbind(M,M1))
如果是芯片表达矩阵,因为样本数量太多,而且分组很复杂,所以相关性聚类结果会没那么简单。
library(ALL)
data(ALL)
ALL
## ExpressionSet (storageMode: lockedEnvironment)
## assayData: 12625 features, 128 samples
## element names: exprs
## protocolData: none
## phenoData
## sampleNames: 01005 01010 ... LAL4 (128 total)
## varLabels: cod diagnosis ... date last seen (21 total)
## varMetadata: labelDescription
## featureData: none
## experimentData: use 'experimentData(object)'
## pubMedIds: 14684422 16243790
## Annotation: hgu95av2
array_expr=exprs(ALL)
array_pd=pData(ALL)
tmp=array_pd[,c('sex','BT','mol.biol')]
rownames(tmp)=colnames(array_expr)
M=cor(array_expr)
# 这里的分组非常复杂,所以相关系数并不能完全反应样本本身的分组信息
pheatmap::pheatmap(M,annotation_col = tmp)
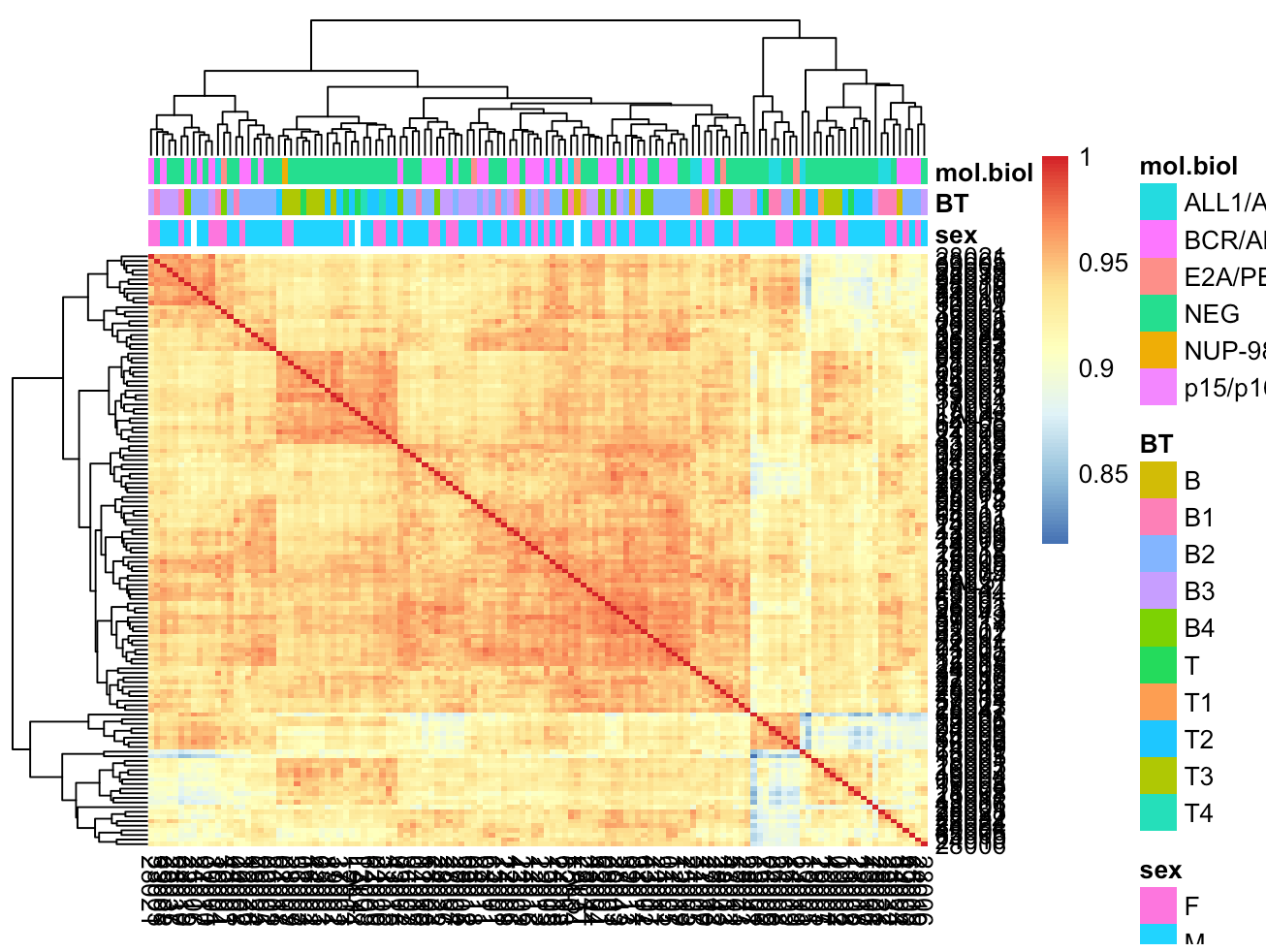
哪怕这个时候,选择同样的策略来挑选高变化的基因。
接下来探索
- 聚类算法
- 层次聚类
- kmeans
- dbscan
- 其它
- 降维算法
- PCA
- t-SNE
- 其它
敬请期待