最近看文献:Integrative Pharmacogenomics Analysis of Patient Derived Xenografts 又一次遇到了codeocean ,算是生物信息学数据分析者的一个福音,因为大量的好文章都是把绘图数据及代码一股脑的打包上去了,关键是都是可以重复出来的!
每个文章的代码都是一个独立的环境
如下所示,code及其配套的data都是独立的文件夹,而且是可以查看的。
可以从中学到其他人的代码技巧,比如判断哪些包没有安装,甚至批量安装R包
cat(sprintf("\n\n##=========== checking if required packages are installed ======\n\n"))
packagesReq <- c("BBmisc", "doParallel", "foreach", "ggplot2", "ggpubr",
"methods", "psych", "reshape2", "Rtsne", "scales", "snow",
"Biobase", "circlize", "ComplexHeatmap", "piano", "PharmacoGx")
packToInst <- setdiff(packagesReq, installed.packages())
if(length(packToInst)>0)
{
cat(sprintf("\nInstalling required packages:\n%s\n", paste0(packToInst, collapse = "\n")))
}
实际上就是一个个独立的docker
docker我们讲解很多次了,具体大家可以浏览我在生信技能树上面写过部分docker教程, 目录如下:
- 用集成了anaconda的docker快速布置生信分析平台
- 我学会docker啦!希望你也可以学会
- 跟着jimmy学docker系列之第2讲:一个软件一个容器
- 跟着jimmy学docker系列之第3讲:为何不创建自己的docker容器呢?
- 跟着jimmy学docker系列之第4讲:docker容器资源调度问题(MAC版本)
- 使用阿里云+Docker分析RNA-Seq与ChIP-Seq
- Docker应用之一键化安装Wordpress(无需代码基础)
- 如何从看不懂Dockerfile到创建自己的镜像
再复习几个docker指令:
docker
docker info ## 可以查看目前机器上面的docker里面有多少容器或者镜像。
docker version
sudo docker search ubuntu
sudo docker run hello-world
## 上面代码下载了一个镜像,启动了一个容器,下面就可以查看它们
sudo docker run ubuntu ## 默认下载最新版docker
docker ps -a ## 查看目前所有没有被销毁的容器进程。
docker images -a ## 查看目前所有的本地镜像
docker volume ls
docker network ls
打开这个codeocean的dockerfile,可以很清楚的看到,就是基于codeocean的r-base:3.4.4-ubuntu16.04这个初始化的空白电脑系统,然后安装几个这篇文章绘图需要的R包,就可以啦!
FROM registry.codeocean.com/codeocean/r-base:3.4.4-ubuntu16.04
ARG DEBIAN_FRONTEND=noninteractive
RUN Rscript -e 'devtools::install_version("BBmisc", \
version = "1.11", \
dependencies = TRUE)'
RUN Rscript -e 'devtools::install_version("Rtsne", \
version = "0.13", \
dependencies = TRUE)'
RUN Rscript -e 'devtools::install_version("doParallel", \
version = "1.0.11", \
dependencies = TRUE)'
RUN Rscript -e 'devtools::install_version("doSNOW", \
version = "1.0.16", \
dependencies = TRUE)'
RUN Rscript -e 'devtools::install_version("foreach", \
version = "1.4.4", \
dependencies = TRUE)'
RUN Rscript -e 'devtools::install_version("ggpubr", \
version = "0.1.7", \
dependencies = TRUE)'
RUN Rscript -e ' \
source("http://bioconductor.org/biocLite.R"); \
biocLite(c( \
"Biobase", \
"ComplexHeatmap", \
"PharmacoGx", \
"piano" \
), suppressUpdates = TRUE)'
RUN Rscript -e 'devtools::install_github("bhklab/Xeva", \
dependencies = TRUE, \
upgrade_dependencies = FALSE, \
ref = "v1.0.0")'
docker的好处就是,随时启动,任意销毁,不需要有任何的心理负担,哪怕你有心理洁癖!
圈圈图
这篇文章展示了一个药物靶点以及其对应的通路关系的圈圈图,如下:
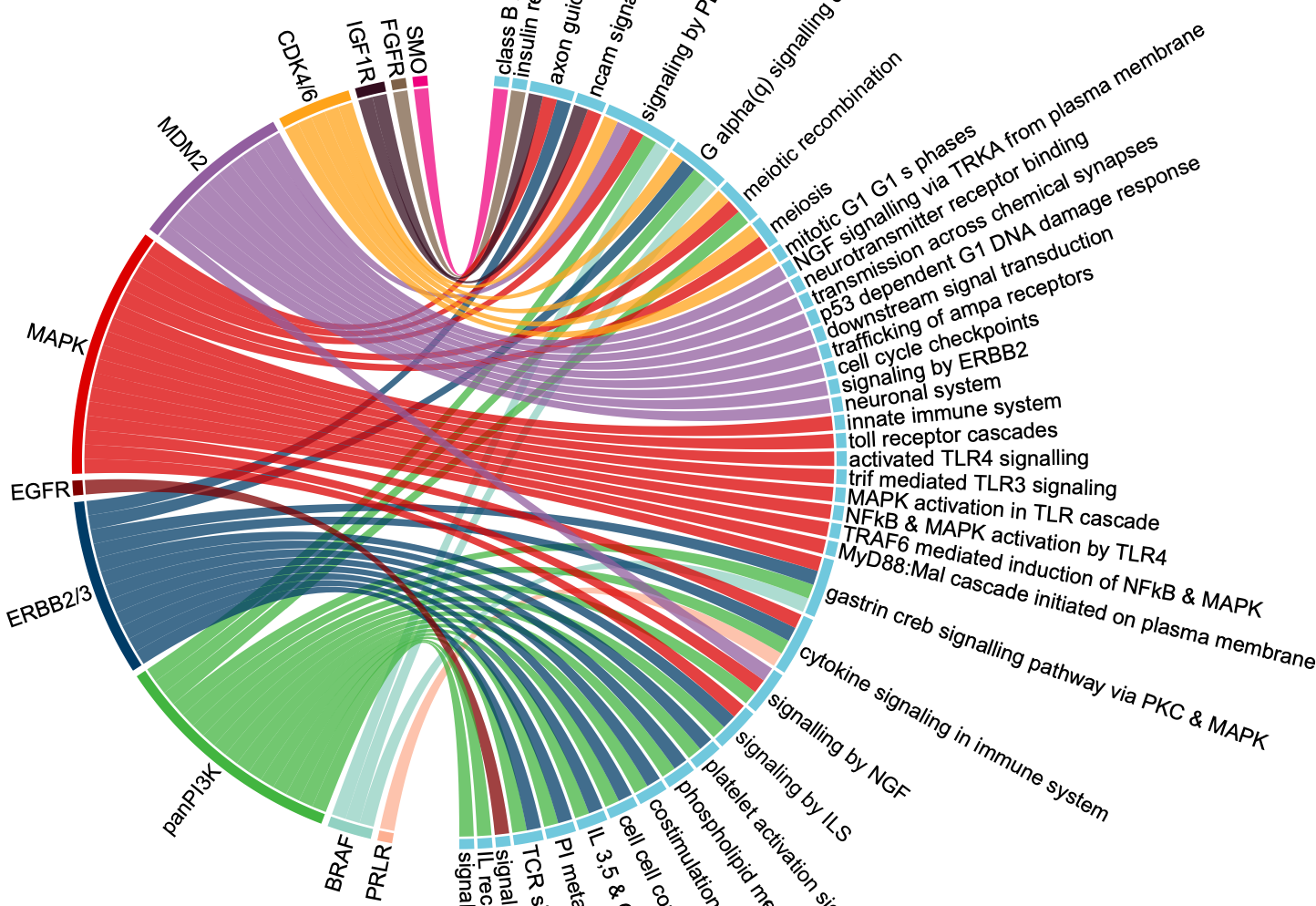
假如你感兴趣绘制这个图的代码,就可以点开看具体的代码实现,如下:
suppressMessages(library(circlize))
library(BBmisc)
library(reshape2)
plot_figure <- function(mat, cirOrd, gap.after, grid.col, colorMat, names2show,
txtCol)
{
paraText <- function(txt, width = 50)
{ paste( strwrap(txt, width = width), collapse = "\n") }
circos.clear()
circos.par(gap.after = gap.after, start.degree = -100,
track.margin = c(0.001, 0.002))
chordDiagram(mat, order = cirOrd, grid.col = grid.col, col=colorMat,
annotationTrack = "grid",
preAllocateTracks = list(track.height= 0.50),
transparency = 0.25)
circos.trackPlotRegion(track.index = 1, panel.fun = function(x, y)
{
xlim = get.cell.meta.data("xlim")
ylim = get.cell.meta.data("ylim")
sector.name = get.cell.meta.data("sector.index")
if(sector.name %in% names2show)
{
circos.text(mean(xlim), ylim[1], sector.name, facing = "clockwise",
niceFacing = TRUE, adj = c(0, 0.5),
col = txtCol[sector.name],cex = 1.5)
}
}, bg.border = NA)
circos.clear()
}
主要是 chordDiagram 和 circos.trackPlotRegion两个函数,来自于circlize这个R包。