最近收到粉丝求助,本来是询问富集分析的时候,我给的参考代码,为什么pvaluecutoff值和qvaluecutoff值设到0.9,其实是怕大家富集不到结果。然后Y叔在自己的微信公众号中提到“富集不到结果才是正确的结果”,采用了更加稳妥和可靠的方法来判断富集结果,而粉丝的数据在DAVID中能有结果,可在Y叔的包里,结果就少了一些,如何决定采取哪个?最后又讨论到DAVID结果可视化,网上资源少,他只能做成条图,需要我给指条方向!
这个时候我还是推荐了Y叔的clusterProfiler ,就去找了找其官网,的确可视化方法又多了几个:
- barplot
- cnetplot
- dotplot
- emapplot
- gseaplot
- goplot
- upsetplot
好几个都是以前没有介绍过的,有趣的是我准备浏览这些可视化函数的帮助文档的时候,看到了这样的话:Please go to https://yulab-smu.github.io/clusterProfiler-book/ for the full vignette.
重点来了,Y叔特意为其包写了一本书来介绍其用法。
镇书之图
打开Y叔的书,映入眼帘的就是流程图:
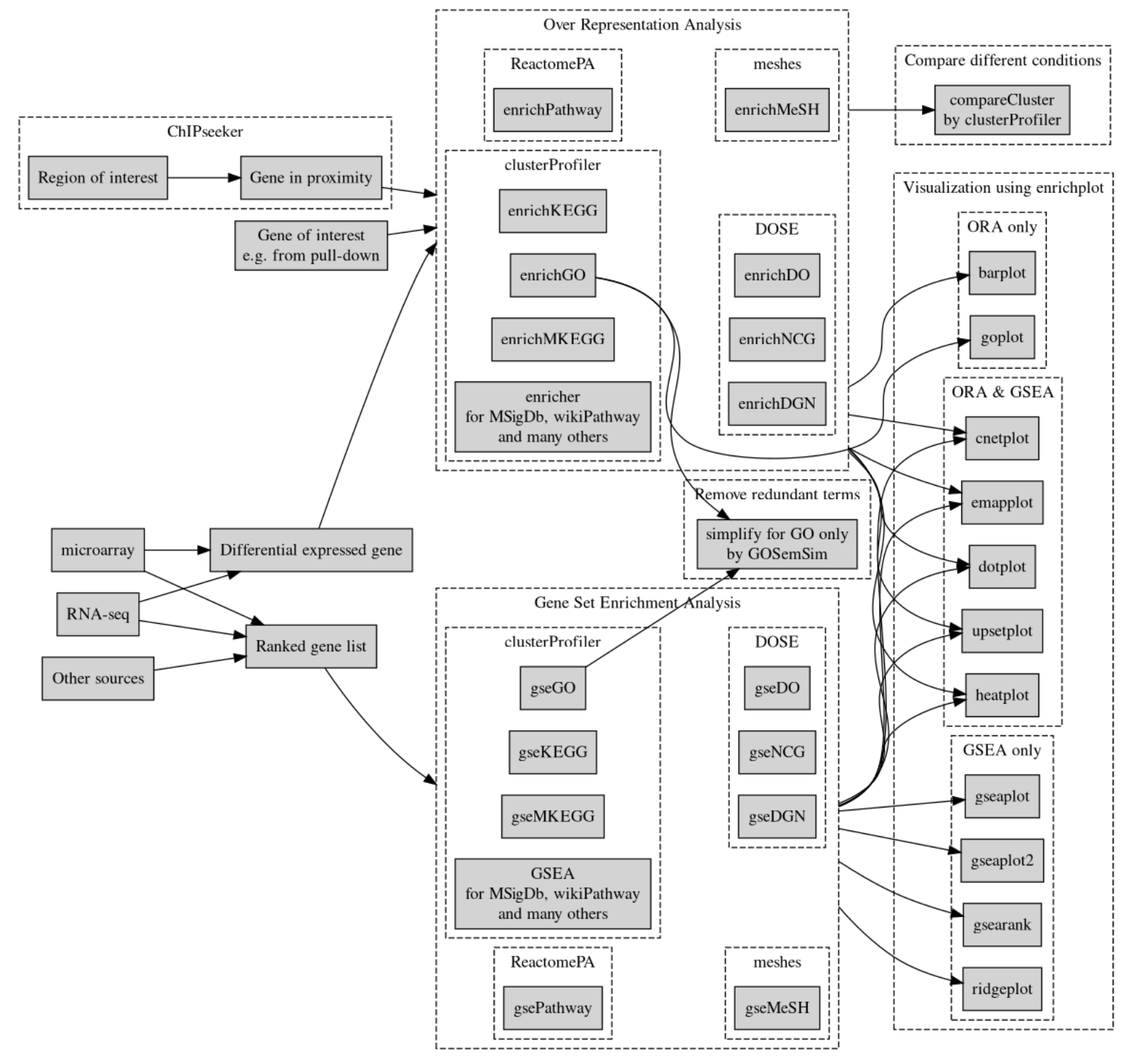
可以说是,跟着这些函数和数据库,都可以入门生物信息学数据分析了。
做完差异分析
GEO数据挖掘代码,很容易得到上下调基因,而且转为ENTREZID,后续分析都以这个为主线。
load(file = 'deg.Rdata')
head(deg)
## 不同的阈值,筛选到的差异基因数量就不一样,后面的超几何分布检验结果就大相径庭。
logFC_t=1.5
deg$g=ifelse(deg$P.Value>0.05,'stable',
ifelse( deg$logFC > logFC_t,'UP',
ifelse( deg$logFC < -logFC_t,'DOWN','stable') )
)
table(deg$g)
head(deg)
deg$symbol=rownames(deg)
library(ggplot2)
library(clusterProfiler)
library(org.Hs.eg.db)
df <- bitr(unique(deg$symbol), fromType = "SYMBOL",
toType = c( "ENTREZID"),
OrgDb = org.Hs.eg.db)
head(df)
DEG=deg
head(DEG)
DEG=merge(DEG,df,by.y='SYMBOL',by.x='symbol')
head(DEG)
save(DEG,file = 'anno_DEG.Rdata')
gene_up= DEG[DEG$g == 'UP','ENTREZID']
gene_down=DEG[DEG$g == 'DOWN','ENTREZID']
或者你提供其它方法,拿到如下的数据,会R语言的都知道是简单的向量罢了。

如果你确实差异分析都不会,就是拿到上下调基因集有困难,看:多分组差异分析续集 里面的教程哈。
最简单的超几何分布检验
这里就拿KEGG数据库举例吧,拿自己判定好的上调基因集进行超几何分布检验,如下:
gene_down
gene_up
enrichKK <- enrichKEGG(gene = gene_up,
organism = 'hsa',
#universe = gene_all,
pvalueCutoff = 0.1,
qvalueCutoff =0.1)
head(enrichKK)[,1:6]
browseKEGG(enrichKK, 'hsa04512')
dotplot(enrichKK)
enrichKK=DOSE::setReadable(enrichKK, OrgDb='org.Hs.eg.db',keyType='ENTREZID')
enrichKK
得到的enrichKK是Y叔定义好的对象,这个对象值得大家用心摸索一下,毕竟Y叔后续的可视化函数,都是基于此。
最基础的条形图和点图
其实就是超几何分布检验的结果,如何可视化,毕竟大家都喜欢看图

上面的表格进行可视化,可以是最基础的条形图和点图,使用Y叔的可视化函数即可,作用于其定义好的对象。
#条带图
barplot(enrichKK,showCategory=20)
#气泡图
dotplot(enrichKK)
这个没有啥悬念,虽然也是可以调整一些参数,可视化上面的表格里面的其它结果
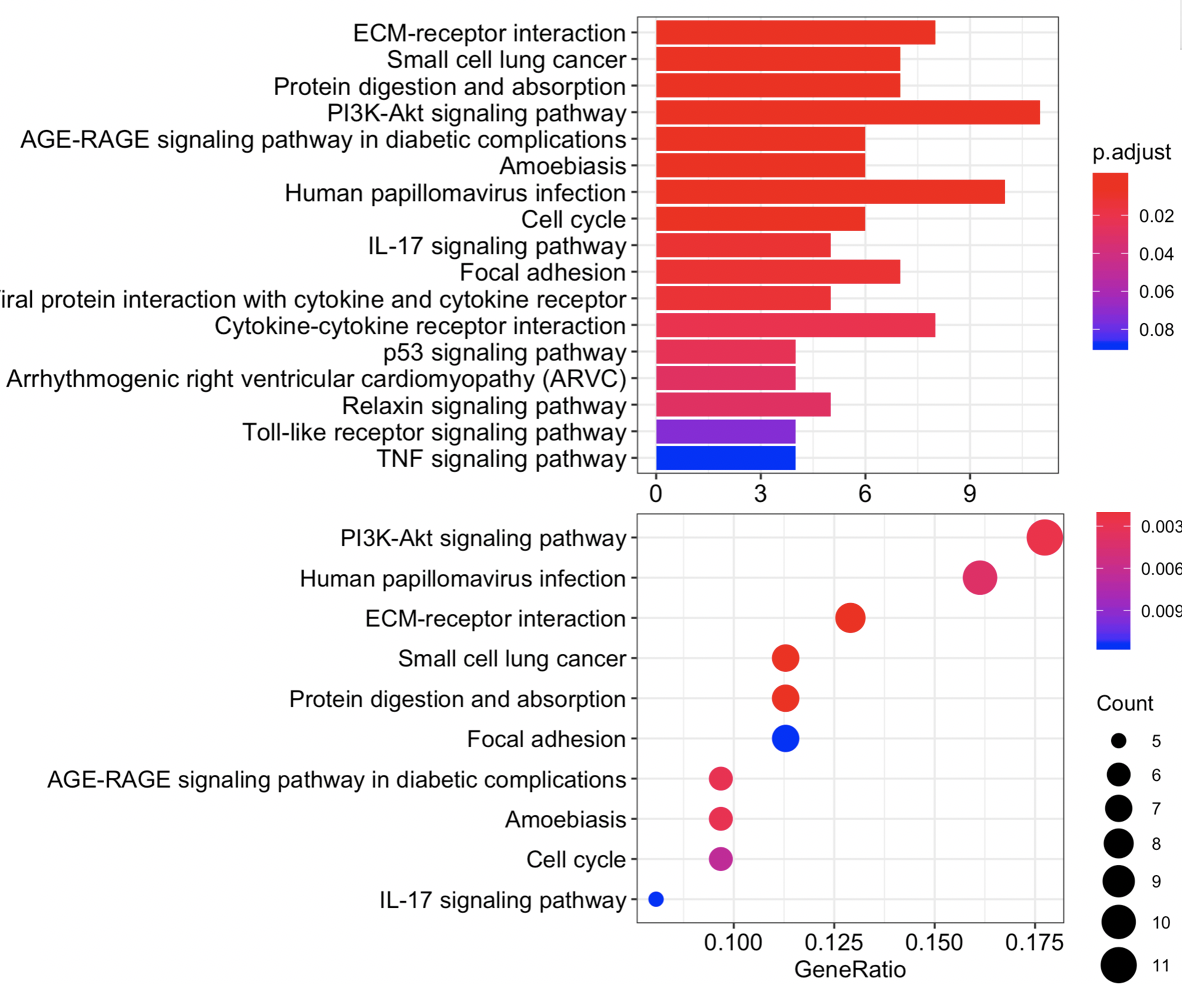
一般来说,我们会喜欢点图一点。
通路与基因之间的关系可视化
这里有两种形式,同一个函数,不同的参数即可:
cnetplot(enrichKK, categorySize="pvalue", foldChange=geneList,colorEdge = TRUE)
cnetplot(enrichKK, foldChange=geneList, circular = TRUE, colorEdge = TRUE)
这个就不好说大家会更喜欢哪个了
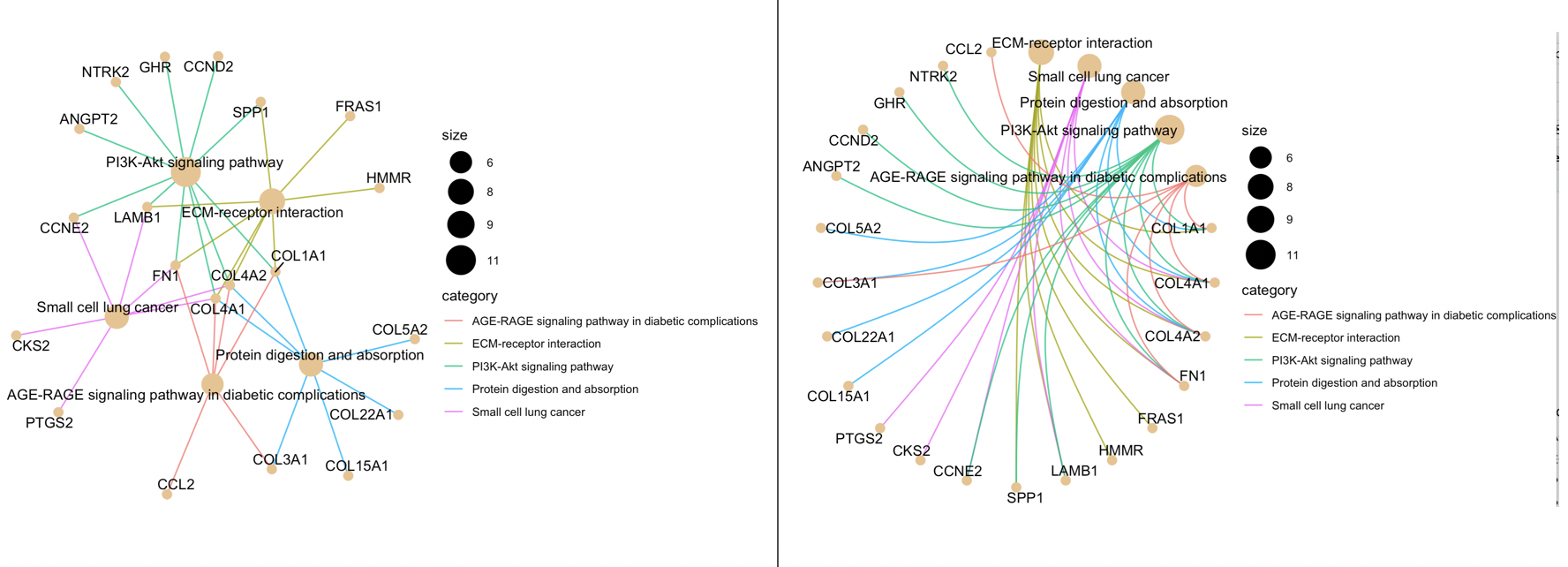
展现的是不同的通路,共有基因情况,可以看到有一些基因是可以属于不同的通路的。
通路与通路之间的连接展示
其实上面的通路与基因之间的关系可视化已经告诉我们了,有一些通路是可以共享一些基因的,这样的话,这些共享基因的通路的相似性就会高一点,它们可以看做是通路模块。
所以就有可视化函数来具体的查看通路与通路之间的连接,不需要展示基因了,代码如下:
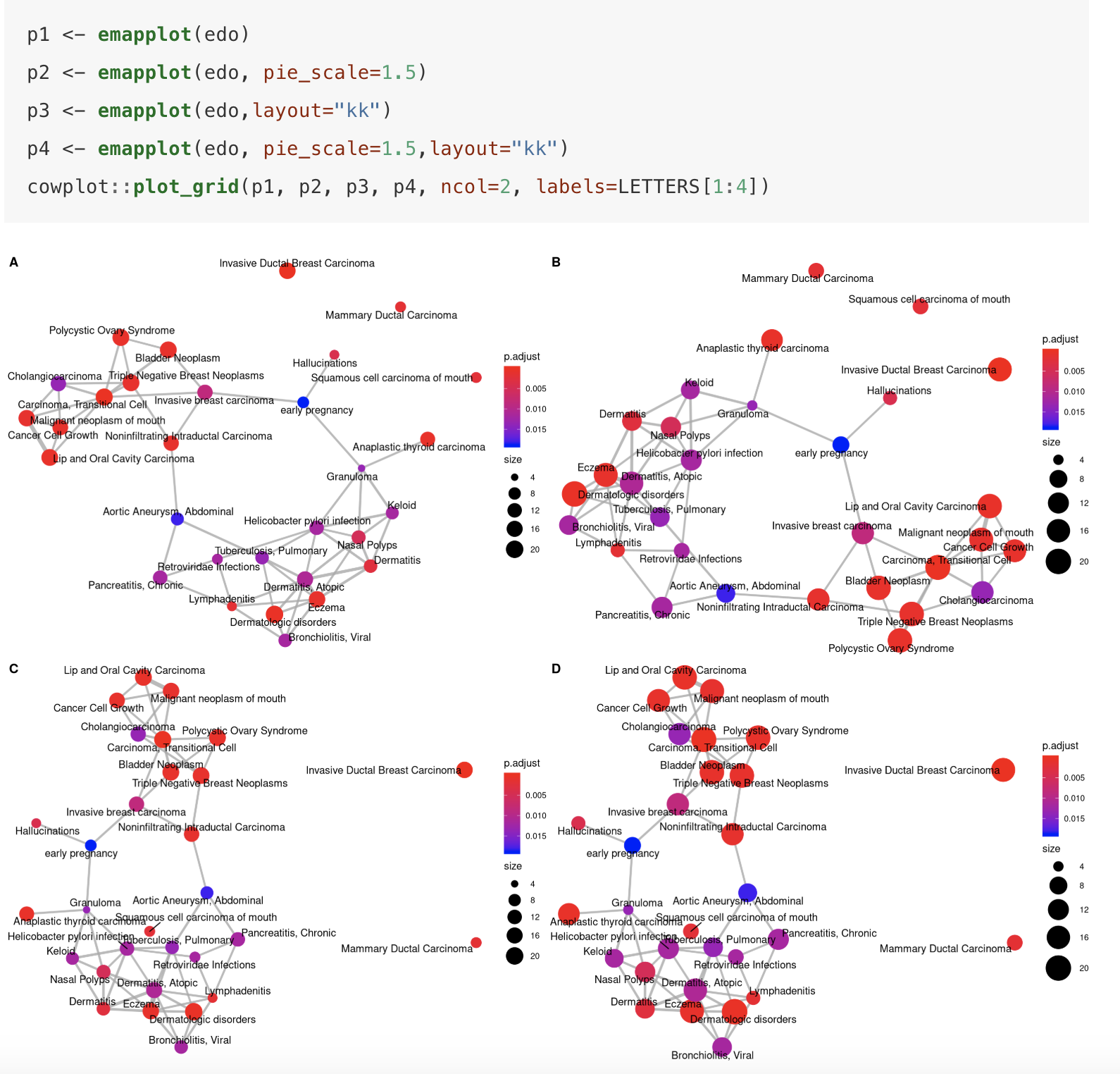
emapplot(enrichKK)
可以看到, p53通路和细胞周期通路,连接起来了,说明它们两个共有基因数量多。
而 toll-like受体,IL-17等,都是免疫相关通路,所以连接起来了。
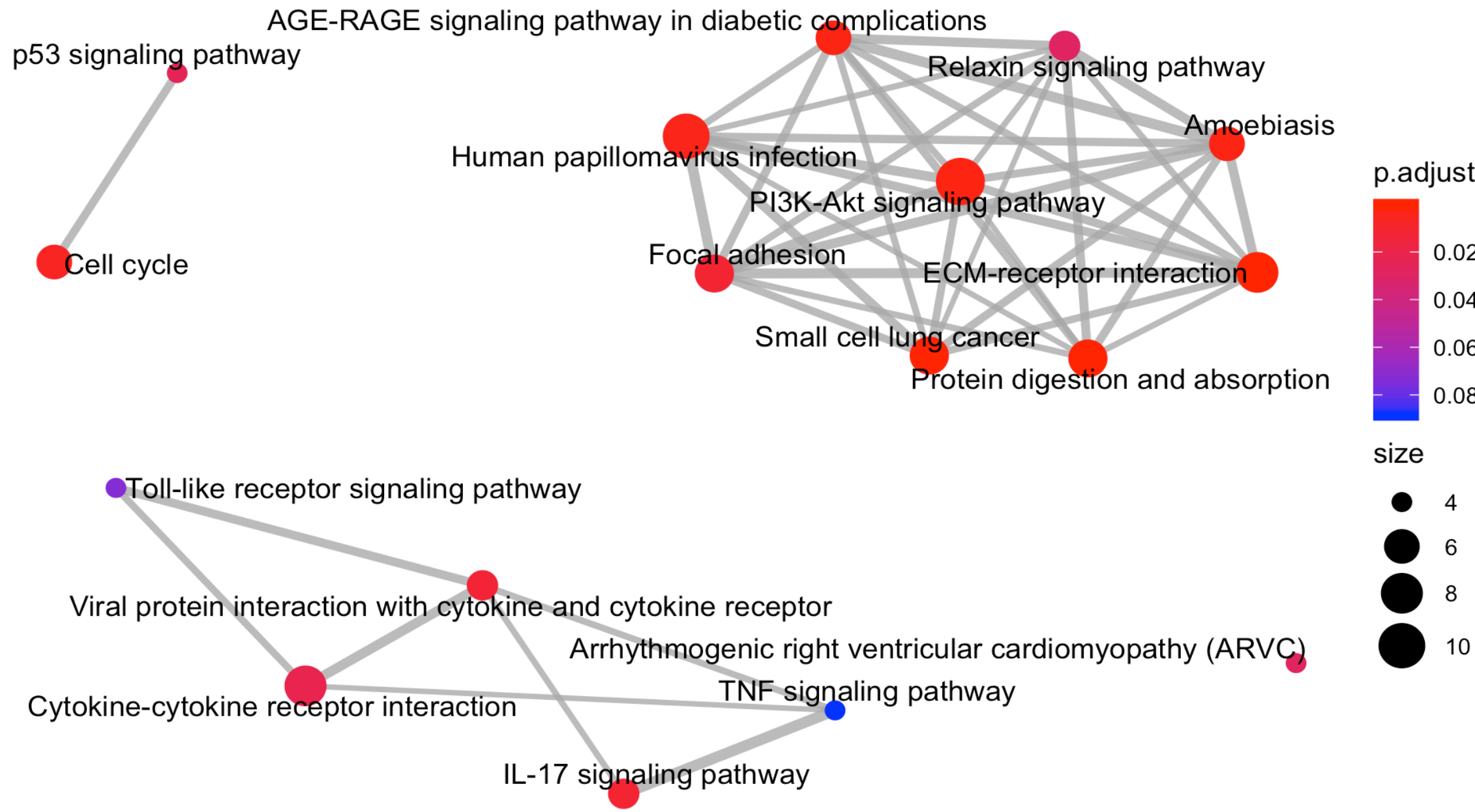
当然了,Y叔自己的例子更漂亮,而且多元化,毕竟参数都是他定义的。
热图展现通路与基因之间的关系
前面的cnetplot就是展现通路与基因之间的关系,但是它的特色的炫酷,其实看起来很烧脑,如果仅仅是为了展现通路与基因之间的关系,热图更靠谱,代码如下:
heatplot(enrichKK)
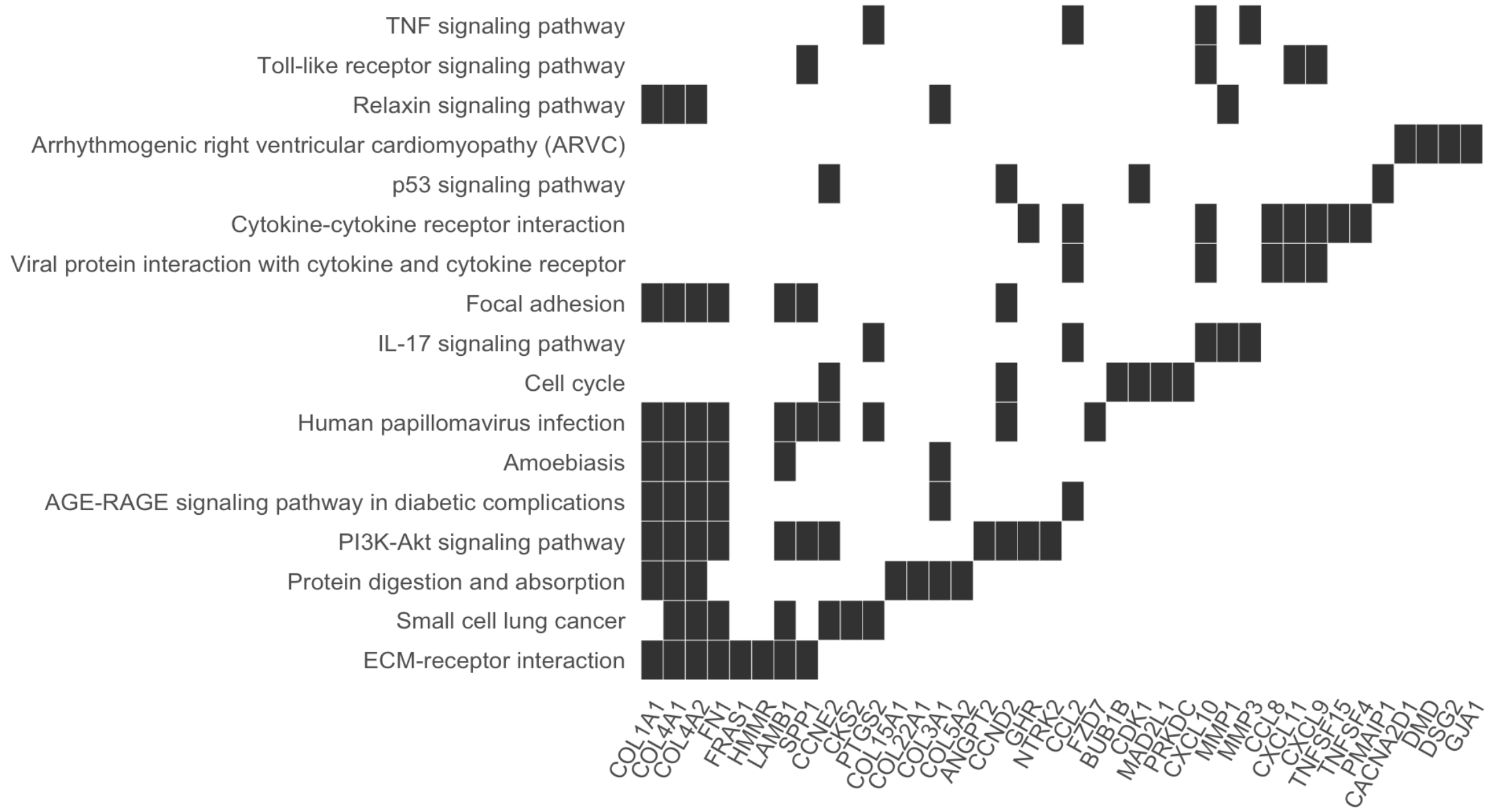
如果你仔细看这个图,就会发现,它不仅仅是通路与基因之间的关系,还包含了通路与通路之间的连接关系,如果两个通路共有的基因比较多,当然在emapplot里面,就会连接在一起。
我个人认为,其实 heatplot是最强大的,但是呢, 没有cnetplot和emapplot炫酷,而barplot和dotplot就太朴素了。最后,如果你是做GO数据库呢,其实还有一个goplot可以试试看,当然是以Y叔的书为主啦。
图都可以可以修饰的
对ggplot语法有所了解的朋友都知道,其产出的图都是可以叠加各种属性的。同理,Y叔的这些可视化函数,也可以继续映射基因的其它属性,比如变化倍数等等。
如果你还不知道Y叔是谁
南方医科大学教授博导,看他的招博士通知吧:常年招硕博士、博士后一起搞生信或被生信搞