昨天宣传了Y叔的clusterProfiler包之让人眼花缭乱的可视化炫技,见:为R包写一本书(向Y叔致敬) 。有趣的是粉丝留言提到:Y叔的图必须以富集出来的对象,比如enrichKK来画,如果是自己准备数据框的话,就不行,不知道作者有没有好的建议?
实际上,我很能理解粉丝的心情, 确实就这么一个数据集,使用Y叔的clusterProfiler包的结果不满意,就不得不使用在线网页工具:Functional Annotation Tool DAVID Bioinformatics Resources 6.8, NIAID/NIH ,反正也是是可以做GO/KEGG数据库注释(通常就是超几何分布检验啦)的,只需要用户上传自己拿的的基因集就可以,大大的方便了生物学家对数据库的使用。
但是DAVID工具拿到的是分析结果,导入R也是一个数据框,的确不符合Y叔各种可视化函数的输入要求。这个的确大部分初学者是无法解决的,那么我写这个教程就意义重大了。
首先查看Y叔自己的可视化函数要求的对象
具体可视化函数见:为R包写一本书(向Y叔致敬) ,我这里直接使用示例数据啦,代码如下:
library(clusterProfiler)
data(gcSample)
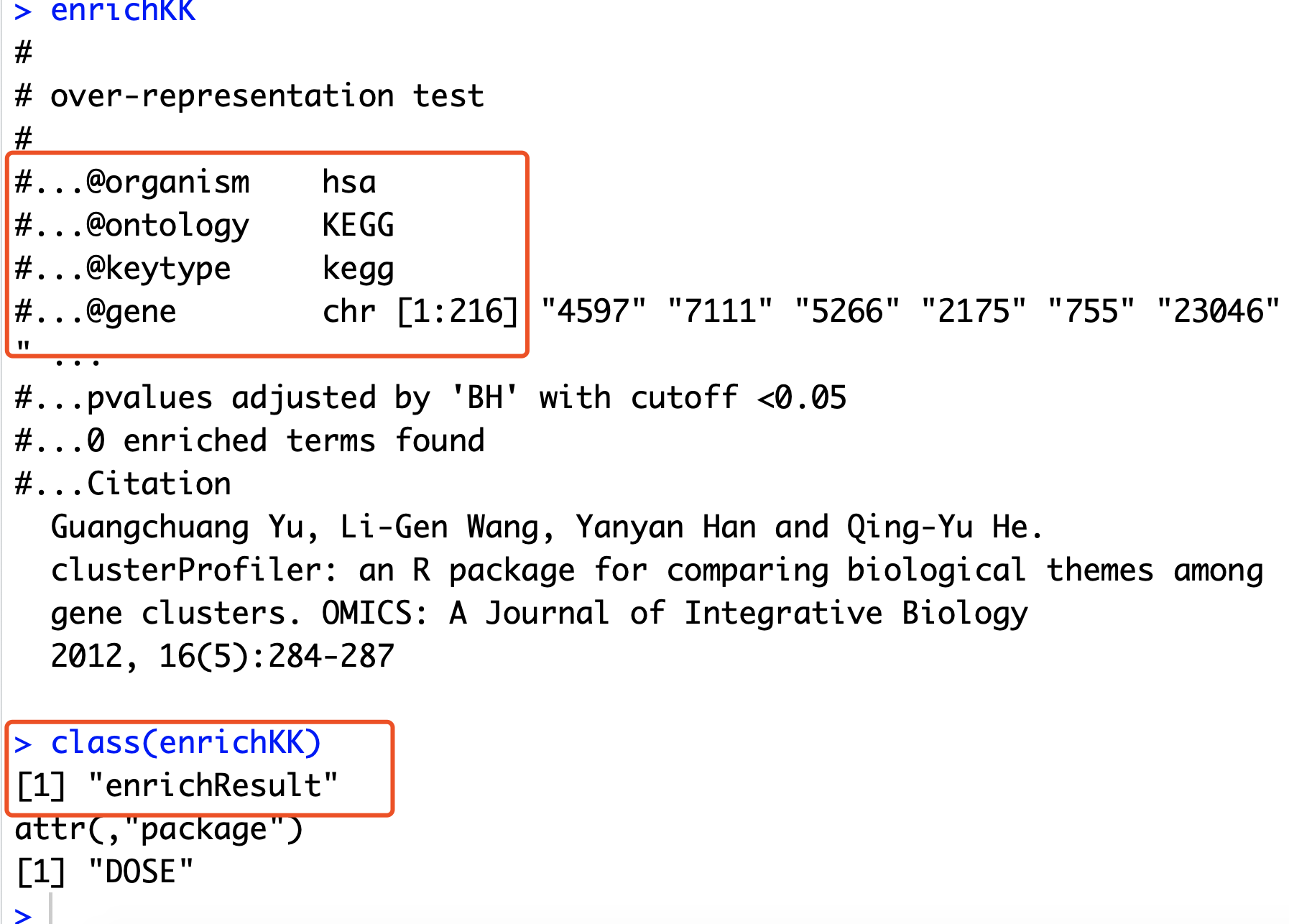
enrichKK <- enrichKEGG(gcSample[[1]])
enrichKK
class(enrichKK)
可以看到,这个enrichResult对象,里面的东西还蛮多的!
这个对象,当然是可以被Y叔自己的可视化函数进行绘图的啦。
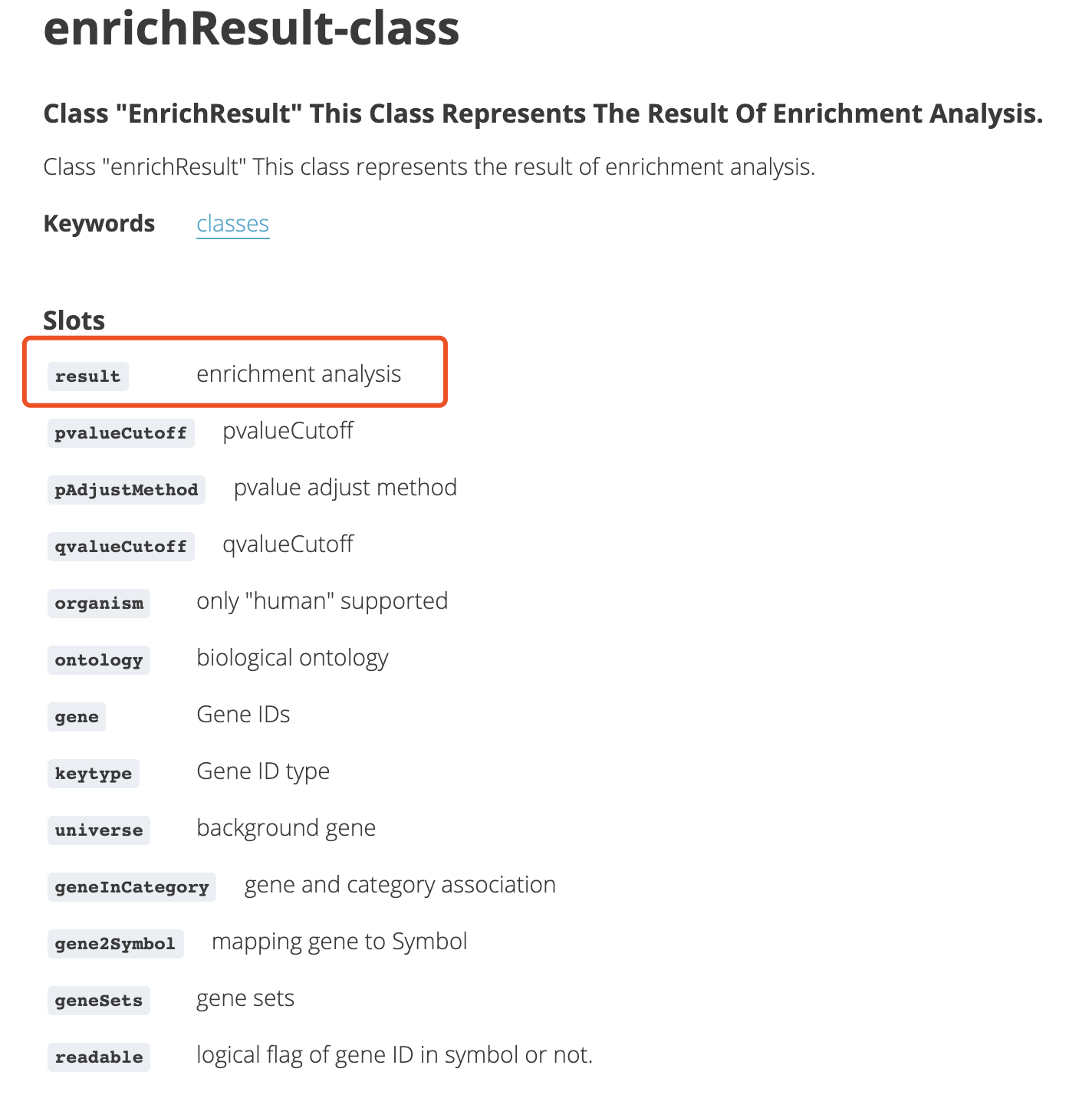
简单谷歌就可以看到该对象的详细信息:https://www.rdocumentation.org/packages/DOSE/versions/2.10.6/topics/enrichResult-class
但是我们提到过,后续的可视化函数,其实重要的就是超几何分布检验后的结果表格而已,你采用DAVID工具拿到的是分析结果,导入R也是一个数据框,并没有本质上区别。
但是数据框毕竟是没办法被可视化函数识别的啊!
> dotplot(enrichKK@result)
Error in (function (classes, fdef, mtable) :
unable to find an inherited method for function ‘dotplot’ for signature ‘"data.frame"’
毫无疑问的报错!
解决方案其实很简单,把你的数据框转为Y叔自己的可视化函数所需要的这个enrichResult对象,虽然里面的东西很多,但是你只需要包超几何分布检验后的结果表格是对的即可。
x=enrichKK@result
head(x[,1:4])
> colnames(x)
[1] "ID" "Description" "GeneRatio" "BgRatio" "pvalue" "p.adjust" "qvalue"
[8] "geneID" "Count"
这样的结果表格

最好是保证列数目一样哦。
创建S4对象
的大部分人来说,S4对象是绝对的超纲啦,不过,这里我只需要你记住两个函数,而且只需要使用一个函数即可。定义一个S4对象,使用函数setClass ,实例化一个,使用函数new 。
y=new("enrichResult",
result=x)
y
dotplot(y,orderBy ='GeneRatio')
虽然这个时候,我们创造的y这个假冒伪劣的enrichResult对象甚至是错的,但是神奇的事情发生了,出图啦!
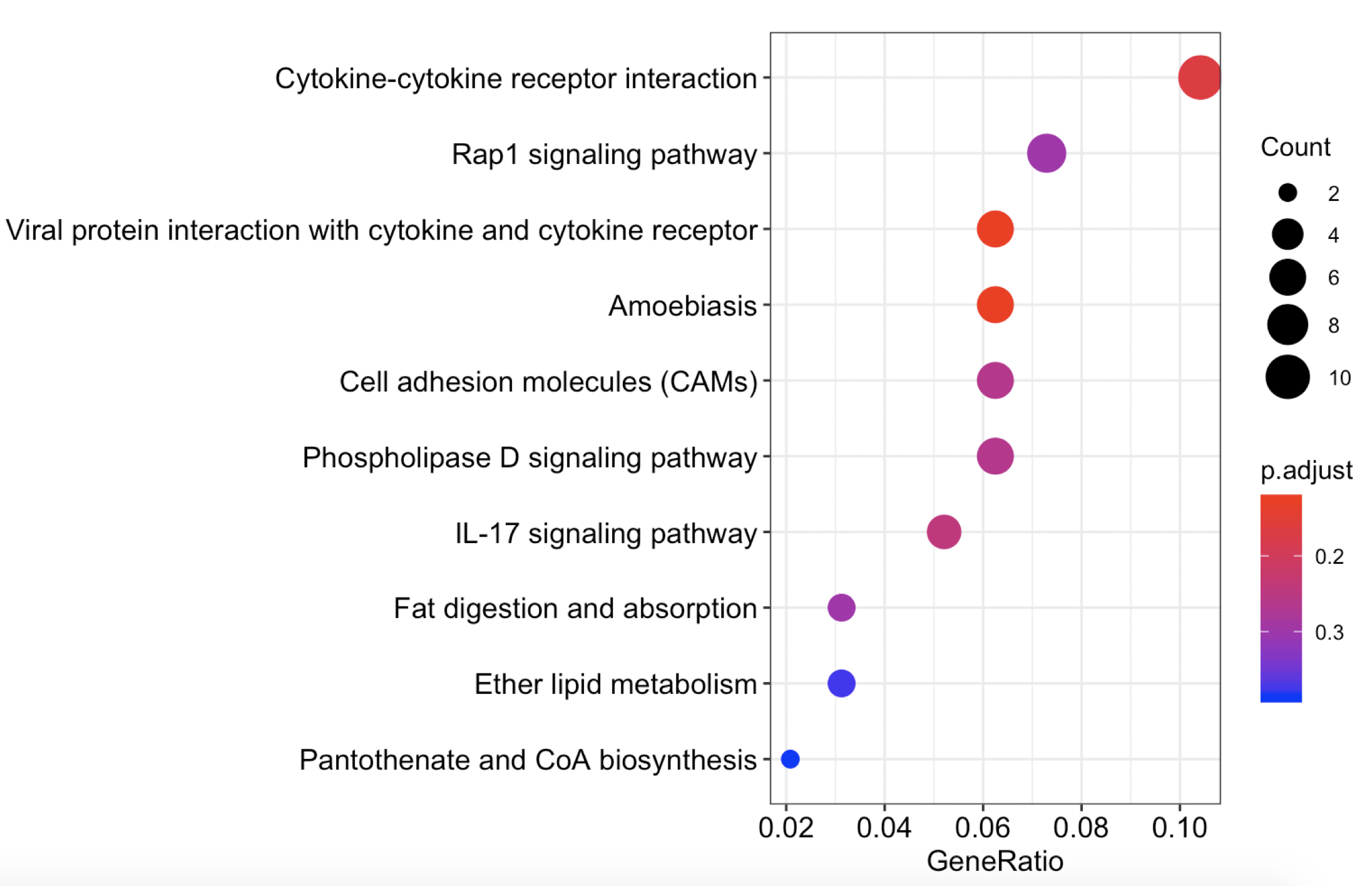
是不是很激动啊!
其它对象同理也可以自由创造
你把下面的代码运行一波就明白了,其实就是需要花点时间了解一下作者定义好的对象,然后把相应的数据准备好,使用函数new 构造一下即可。
library(clusterProfiler)
data(gcSample)
x <- compareCluster(gcSample,fun = "enrichKEGG")
p <- dotplot(x, showCategory=10)
p
x
class(x)
# https://www.rdocumentation.org/packages/clusterProfiler/versions/3.0.4/topics/compareClusterResult-class
# compareClusterResult:'data.frame': 56 obs. of 10 variables:
tmp=as.data.frame(x)
# 这里是S4对象,需要掌握两个函数:
# 定义一个S4对象,使用函数setClass
# 实例化一个,使用函数new
y=new("compareClusterResult",
compareClusterResult=as.data.frame(x),
fun='enrichGO')
y
class(y)
dotplot(y, showCategory=10)
每个对象都有它的重点,比如上面的 compareClusterResult ,重点就是多个基因集的结果数据框,如下:
> head(tmp[,1:4])
Cluster ID Description GeneRatio
1 X2 hsa04110 Cell cycle 18/377
2 X2 hsa05169 Epstein-Barr virus infection 23/377
3 X2 hsa05340 Primary immunodeficiency 8/377
4 X3 hsa04512 ECM-receptor interaction 9/184
5 X3 hsa04060 Cytokine-cytokine receptor interaction 17/184
6 X3 hsa04151 PI3K-Akt signaling pathway 19/184
> colnames(tmp)
[1] "Cluster" "ID" "Description" "GeneRatio" "BgRatio" "pvalue" "p.adjust"
[8] "qvalue" "geneID" "Count"
>