虽然说我的大部分教程是针对人类或者小鼠的数据分析,实际上这些分析很容易迁移到其它物种,可能比较麻烦的就是最上游的关于参考基因组和注释文件信息的选择吧,这里讲一下猫猫狗狗的数据分析吧!
首先搜索了解物种基础知识
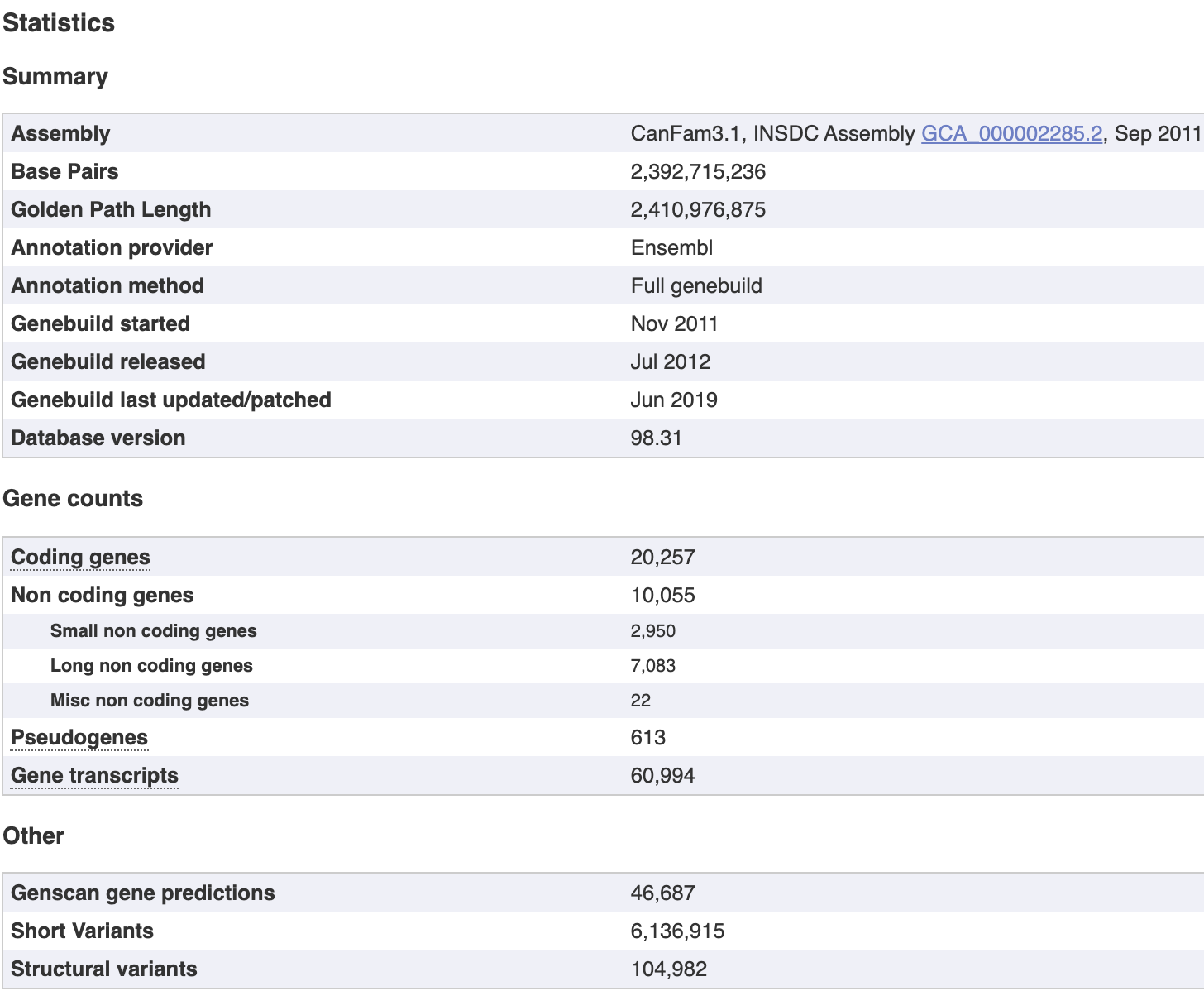
比如搜索dog的:Canis lupus familiaris - Ensembl genome browser 98 就拿到了家犬的参考基因组,实际上狗这个物种本身非常复杂,丰富多彩!如果你看到不同品种的狗狗寿命排行榜,你会崩溃:
(平均寿命,仅供参考)
迷你贵宾犬 14.8岁
拉布来多猎犬 12.6岁
玩具贵宾犬 14.4岁
美国可卡 12.5岁
迷你腊肠犬 14.4岁
柯利牧羊犬 12.3岁
惠比特犬 14.3岁
阿富汗猎犬 12.0岁
松狮犬 13.5岁
金毛寻回猎犬 12.0岁
西施犬 13.4岁
英国可卡 11.8岁
比格猎兔犬13.3岁
爱尔兰雪达 11.8岁
北京犬 13.3岁
威尔士柯基 11.3岁
喜乐帝犬 13.3岁
萨摩耶犬 11.0岁
边境牧羊犬 13.0岁
拳师犬 10.4岁
吉娃娃犬 13.0岁
德国牧羊犬 10.3岁
猎狐梗犬 13.0岁
杜宾犬 9.8 岁
巴基度犬 12.8岁
大丹犬 8.4 岁
西高地白梗犬 12.8岁
伯恩山犬 7.0 岁
约克夏 12.8岁
此外串种的寿命在 12.6 岁
但是我们怎么可能为各个品种的狗都构建参考基因组呢?统一使用ensembl数据库的即可,常见物种都在:http://asia.ensembl.org/info/data/ftp/index.html
The CanFam3.1 assembly was submitted by Dog Genome Sequencing Consortium on November 2011. The assembly is on chromosome level, consisting of 27,106 contigs assembled into 3,310 scaffolds. From these sequences, 39 chromosomes have been built. The N50 size is the length such that 50% of the assembled genome lies in blocks of the N50 size or longer. The N50 length for the contigs is 267,478 while the scaffold N50 is 45,876,610.
值得一提的是有一个 The NHGRI Dog Genome Project 供大家补充阅读:https://research.nhgri.nih.gov/dog_genome/
然后在ensembl数据库下载参考基因组的fa文件
在:ftp://ftp.ensembl.org/pub/release-98/fasta/canis_familiaris/dna/
链接地址是:ftp://ftp.ensembl.org/pub/release-98/fasta/canis_familiaris/dna/Canis_familiaris.CanFam3.1.dna.toplevel.fa.gz
下载的你的linux服务器即可
mkdir -p ~/reference/genome/
cd ~/reference/genome/
mkdir dog_CanFam3.1
cd dog_CanFam3.1
wget ftp://ftp.ensembl.org/pub/release-98/fasta/canis_familiaris/dna/Canis_familiaris.CanFam3.1.dna_rm.toplevel.fa.gz
gunzip -d Canis_familiaris.CanFam3.1.dna_rm.toplevel.fa.gz
# 需要注意文件大小,以及参考基因组是否下载成功哦!
## 2.3G Jan 14 15:50 Canis_familiaris.CanFam3.1.dna_rm.toplevel.fa
然后构建star的索引
可以自由选择不同版本的star软件,或者conda直接傻瓜式安装最新版!
# Get latest STAR source from releases
mkdir -p ~/biosoft/STAR/
cd ~/biosoft/STAR/
wget https://github.com/alexdobin/STAR/archive/2.7.3a.tar.gz
tar -xzf 2.7.3a.tar.gz
cd STAR-2.7.3a
# 也可以选择其它版本的star软件
当然了,你需要自行搜索了解star软件的用法!
mkdir -p ~/reference/index/star/
cd ~/reference/index/star/
mkdir dog_CanFam3.1
cd dog_CanFam3.1
# gtf文件也需要下载,star软件构建index时候需要
wget ftp://ftp.ensembl.org/pub/release-98/gtf/canis_familiaris/Canis_familiaris.CanFam3.1.98.chr.gtf.gz
gunzip -d Canis_familiaris.CanFam3.1.98.chr.gtf.gz
## 需要注意文件大小,以及gtf是否下载成功哦!
## 486M Jan 14 15:49 Canis_familiaris.CanFam3.1.98.chr.gtf
# 文件夹存储我们待会构建好的index
mkdir -p ~/reference/index/star/dog_CanFam3.1/dog
~/biosoft/STAR/STAR-2.7.3a/bin/Linux_x86_64/STAR --runMode genomeGenerate \
--genomeDir ~/reference/index/star/dog_CanFam3.1/dog \
--genomeFastaFiles ~/reference/genome/dog_CanFam3.1/Canis_familiaris.CanFam3.1.dna_rm.toplevel.fa \
--sjdbGTFfile Canis_familiaris.CanFam3.1.98.chr.gtf \
--sjdbOverhang 149 --runThreadN 4
# 如果你的服务器性能不够,不需要设置4个线程哈
得到的的索引文件夹内容如下:
3.1G Jan 14 16:23 Genome
11G Jan 14 16:23 SA
1.5G Jan 14 16:23 SAindex
18K Jan 14 15:58 chrLength.txt
44K Jan 14 15:58 chrName.txt
62K Jan 14 15:58 chrNameLength.txt
36K Jan 14 15:58 chrStart.txt
20M Jan 14 16:19 exonGeTrInfo.tab
8.8M Jan 14 16:19 exonInfo.tab
1.3M Jan 14 16:19 geneInfo.tab
847 Jan 14 16:23 genomeParameters.txt
6.2M Jan 14 16:19 sjdbInfo.txt
6.0M Jan 14 16:19 sjdbList.fromGTF.out.tab
4.8M Jan 14 16:19 sjdbList.out.tab
3.9M Jan 14 16:19 transcriptInfo.tab
log日志里面有时间信息:
Finished loading and checking parameters
Jan 14 15:57:51 ... starting to generate Genome files
Jan 14 16:23:59 ..... finished successfully
DONE: Genome generation, EXITING
可以看到是半个小时左右,当然了,这个速度取决于自己服务器性能哦!
其它软件的索引构建方法类似! 你们可以自己做一下hisat2的。
给你一个作业
同样的流程,下载猪的参考基因组,并且构建star还有hisat2软件的索引哈!