前面我们已经介绍过circRNA的基础概念: 首先了解一下circRNA背景知识,背景知识,以及 circRNA芯片分析的一般流程,但是跟mRNA一样,不仅仅是芯片可以检测,也是可以使用NGS技术,就是circRNA-seq咯。
这里我们一起读文献:Circular RNAs expression profiles in plasma exosomes from early‐stage lung adenocarcinoma and the potential biomarkers 来看看cirRNA-seq分析的一般流程。
circRNA数据分析流程
重中之重其实就是circRNA的定量,在普通的mRNA-seq或者大家熟知的lncRNA-seq里面,我们都是正常的比对,每个reads都会有基因组坐标,然后跟我们的参考gtf文件进行坐标映射后计数即可。
但是cirRNA不太一样,需要先鉴定出来,然后才能定量,比如本研究采用的是就DCC软件流程,其实2015年12月10日发表在《Nucleic Acid Research》 的 https://www.ncbi.nlm.nih.gov/pubmed/26657634 ,文献提到的5种算法预测得到的结果差别较大,而且有很高的假阳性,所以作者建议可以使用多种方法联合预测或者采用去线性RNA建库的方法进行circRNA研究。

首先看circRNA的注释分类
前面我们提到过,circRNA的注释很丰富了,circRNA检测的基本原理是去识别反向剪切的位点(backsplice),最主要的circRNA类型是外显子来源的,当然,在内含子、间区、UTR区域、lncRNA区域以及已知转录本的反义链区域也都鉴定到circRNA,同一个位点可能形成多个circRNA,每个circRNA可能包含一个或多个外显子。
比如本研究鉴定到的两万多个circRNA注释后分类如下;
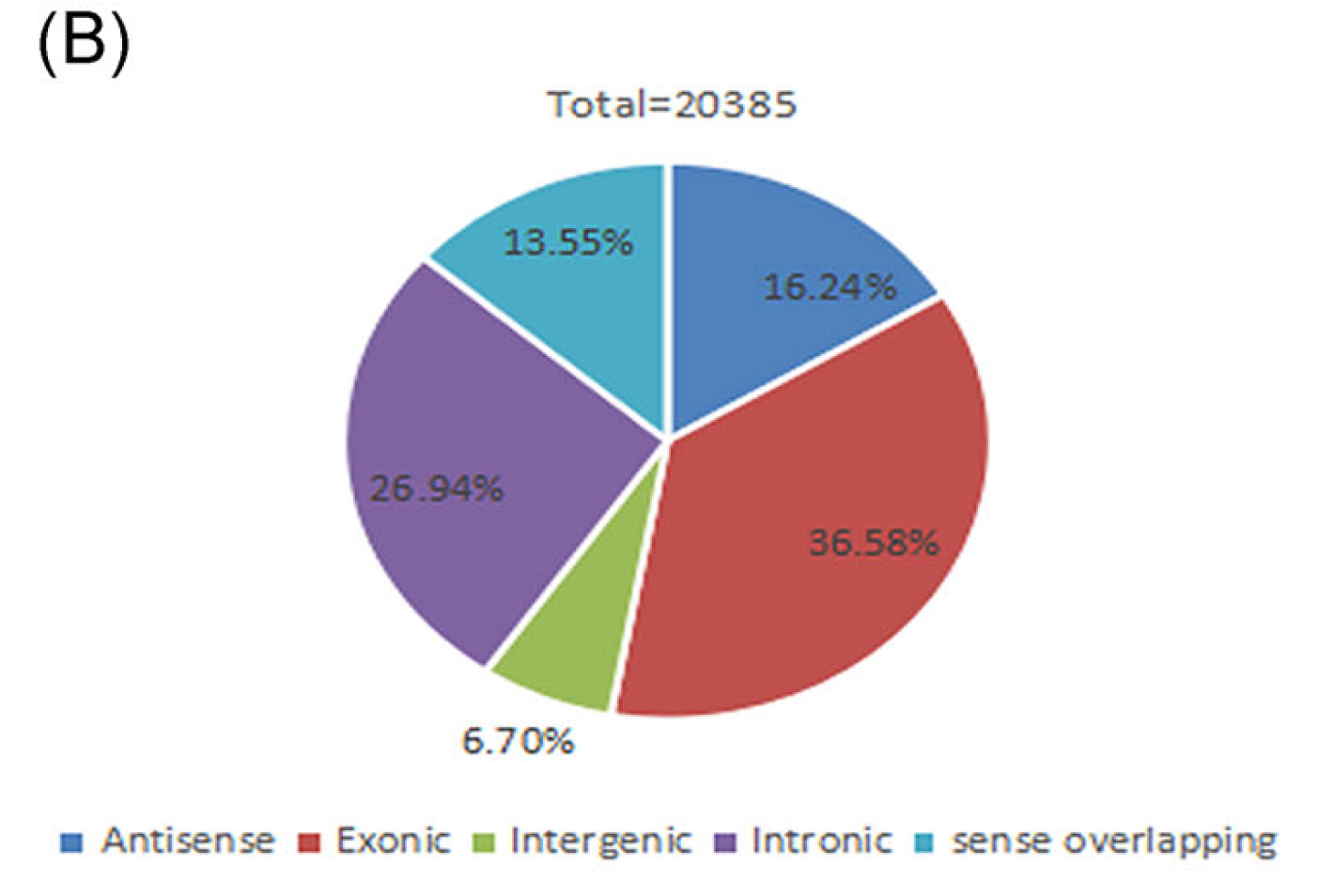
其实本研究还统计了circRNA在不同染色体的数量,还有其它数据库收录与否等等
表达矩阵的标准的差异分析
其实circRNA-seq和circRNA-array最后都是得到表达矩阵,然后就是走标准分析流程,火山图,热图,GO/KEGG数据库注释等等。这些流程的视频教程都在B站和GitHub了,目录如下:
- 第一讲:GEO,表达芯片与R
- 第二讲:从GEO下载数据得到表达量矩阵
- 第三讲:对表达量矩阵用GSEA软件做分析
- 第四讲:根据分组信息做差异分析
- 第五讲:对差异基因结果做GO/KEGG超几何分布检验富集分析
- 第六讲:指定基因分组boxplot指定基因list画热图
仅仅是最后得到的差异分子,并不是以前的mRNA后面的基因名,而是miRNA,lncRNA,甚至circRNA的ID,看起来很陌生罢了。感兴趣可以细读表达芯片的公共数据库挖掘系列推文 ; - 解读GEO数据存放规律及下载,一文就够
- 解读SRA数据库规律一文就够
- 从GEO数据库下载得到表达矩阵 一文就够
- GSEA分析一文就够(单机版+R语言版)
- 根据分组信息做差异分析- 这个一文不够的
- 差异分析得到的结果注释一文就够
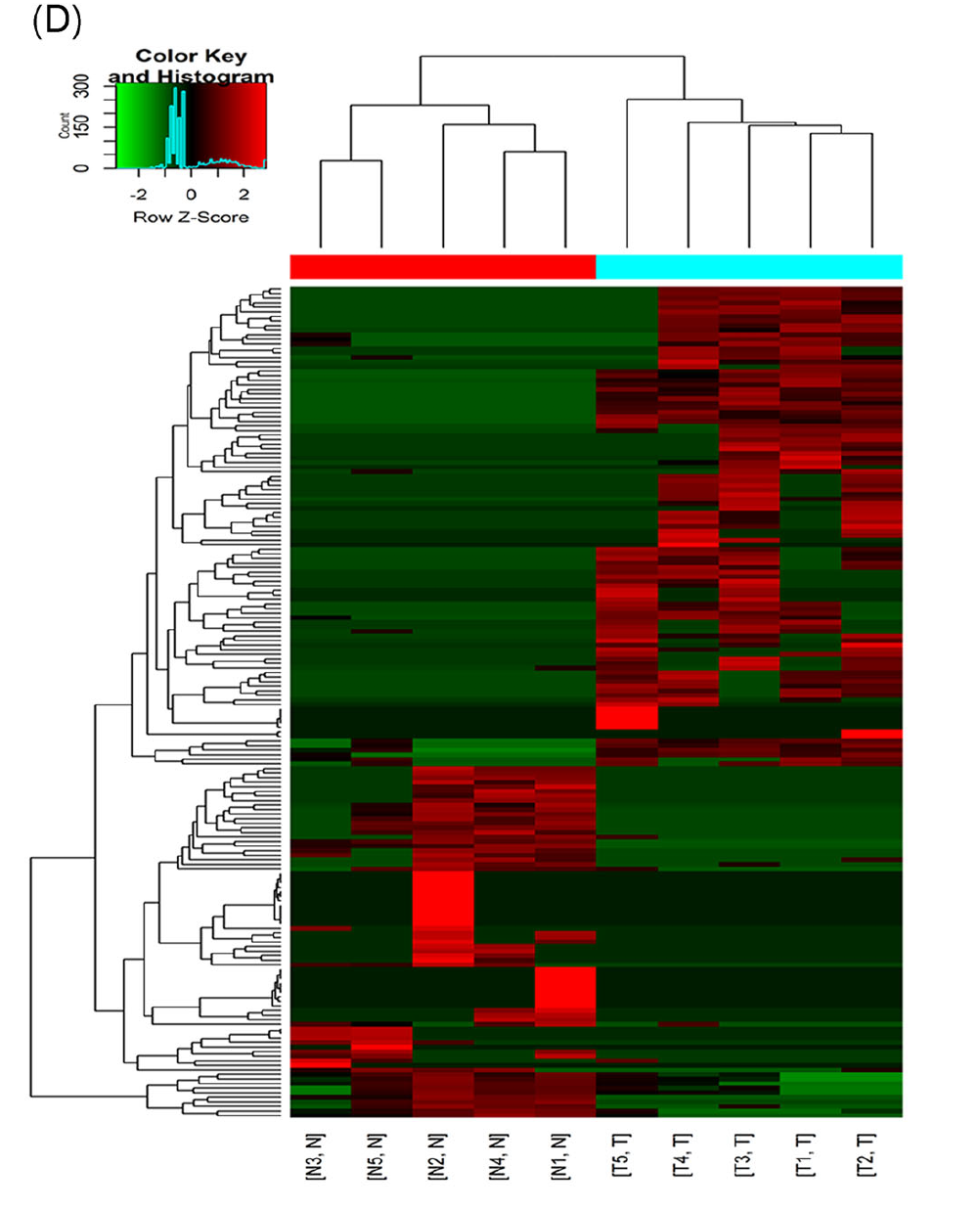
本文得到的热图如下:
也可以列出最显著的那些circRNA的详细信息,如下:
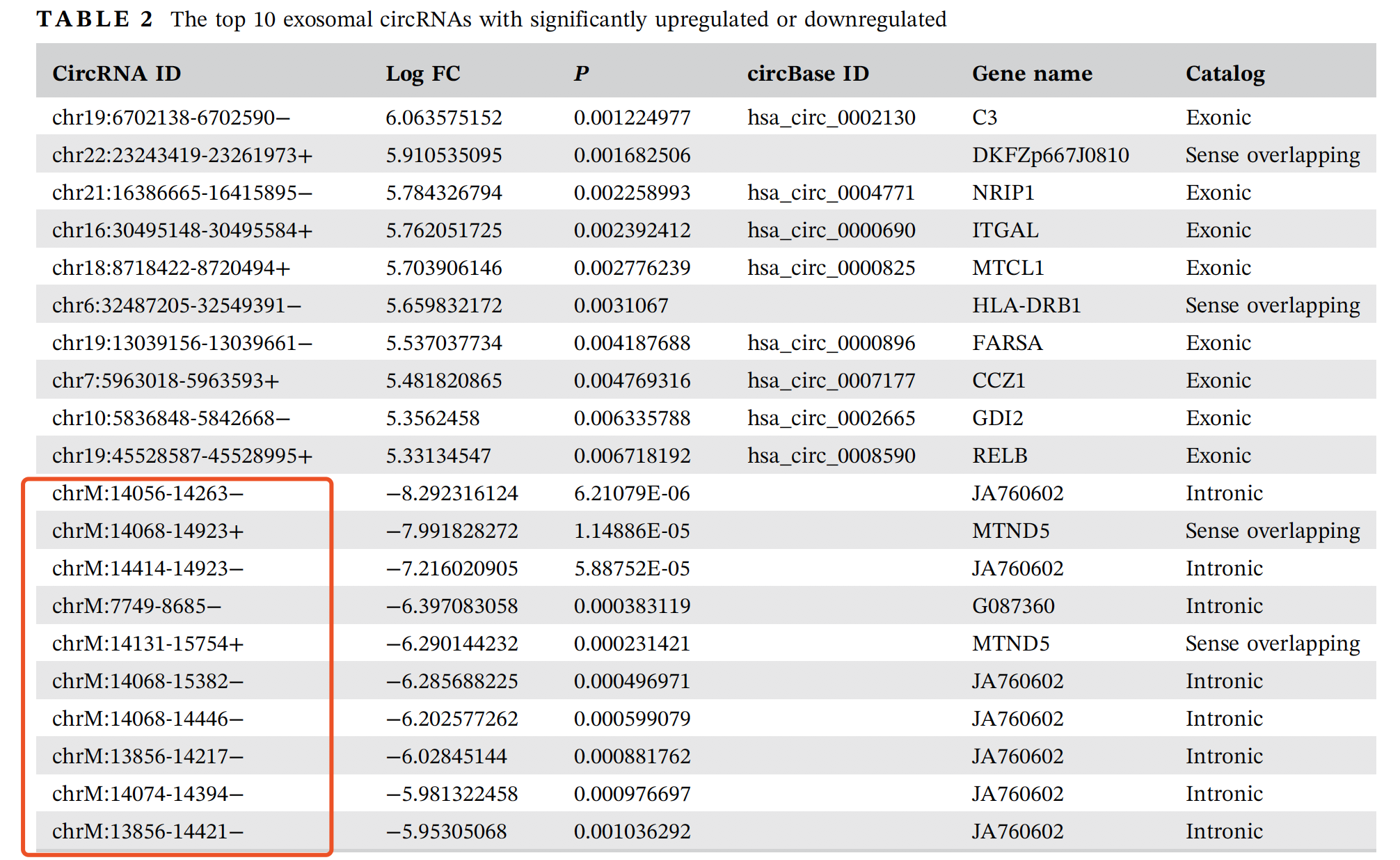
其实这里有一点点问题,看起来线粒体上面的cirRNA有些多了!核心仍然是对circRNA定量和找差异
比如文章:RNA-Seq profiling of circular RNAs in human laryngeal squamous cell carcinomas
