featureCounts我们粉丝都耳熟能详了,我们转录组流程介绍的对比对后的bam文件基于基因注释文件定量的首选软件,用法非常简单,关键是速度飞快,吊打htseq-counts几条街,而用DEXSeq分析可变剪切,外显子差异表达呢,我们以前也分享过用法,那个时候是使用示例的表达矩阵。
回顾一下featureCounts的命令及表达矩阵结果
使用featurecounts时候,我们通常的命令及参数是:
gtf="/home/yb77613/reference/gtf/gencode/gencode.v32.annotation.gtf"
bin_featureCounts="/home/yb77613/biosoft/featureCounts/subread-1.5.3-Linux-x86_64/bin/featureCounts";
$bin_featureCounts -T 4 -p -t exon -g gene_name -a $gtf -o all.counts.id.txt ../hisat2/*.bam 1>counts.id.log 2>&1
实际上,就一个 -t exon -g gene_name 需要理解一下,就是报名数reads数量的时候,只考虑gtf文件里面记录是exon的坐标的reads,然后最后的输出矩阵,以gene_name信息为行。
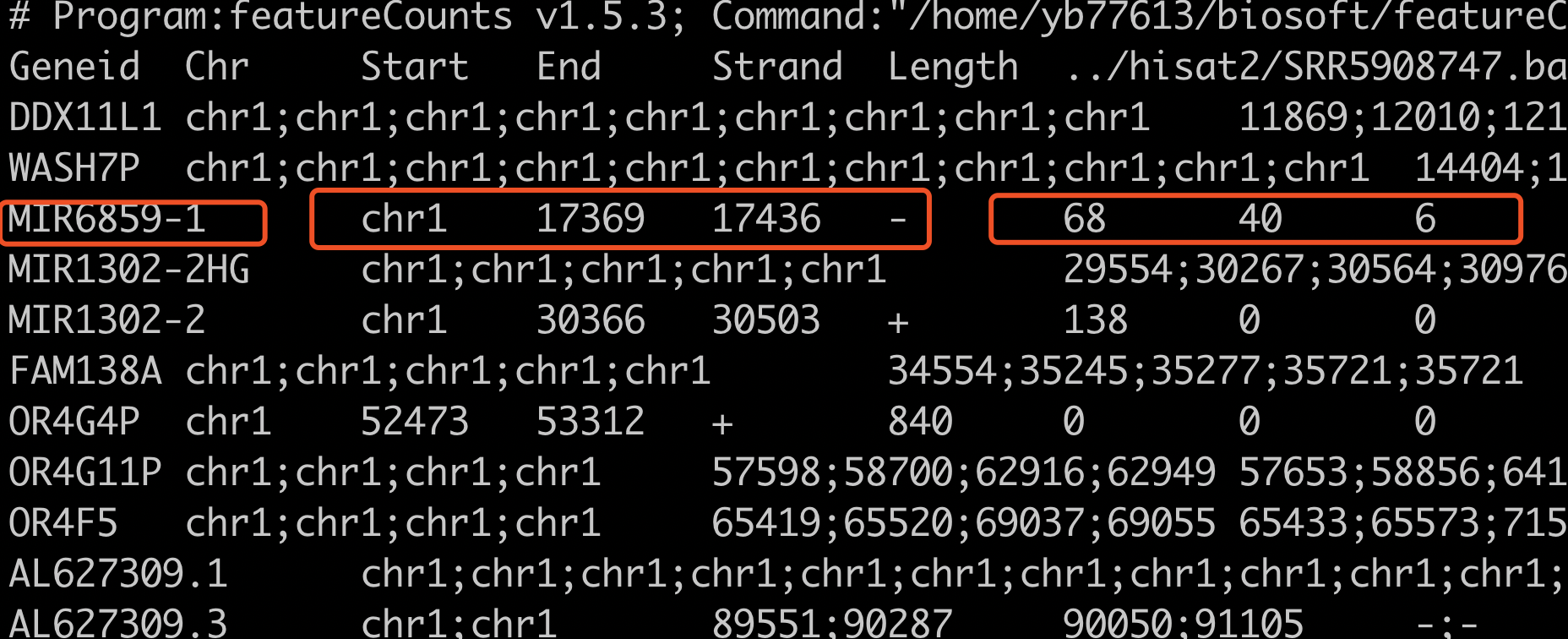
可以很明显的看到,前面的6列,是基因的名字,染色体,起始终止坐标,正负链信息,基因长度信息。我圈起来的那个是miRNA,所以基因长度是68bp。
认识一下DEXSeq的输入表达矩阵
但是使用DEXSeq分析可变剪切,外显子差异表达,需要的不是基于基因的表达矩阵,而是基于exon的,比如官网例子:
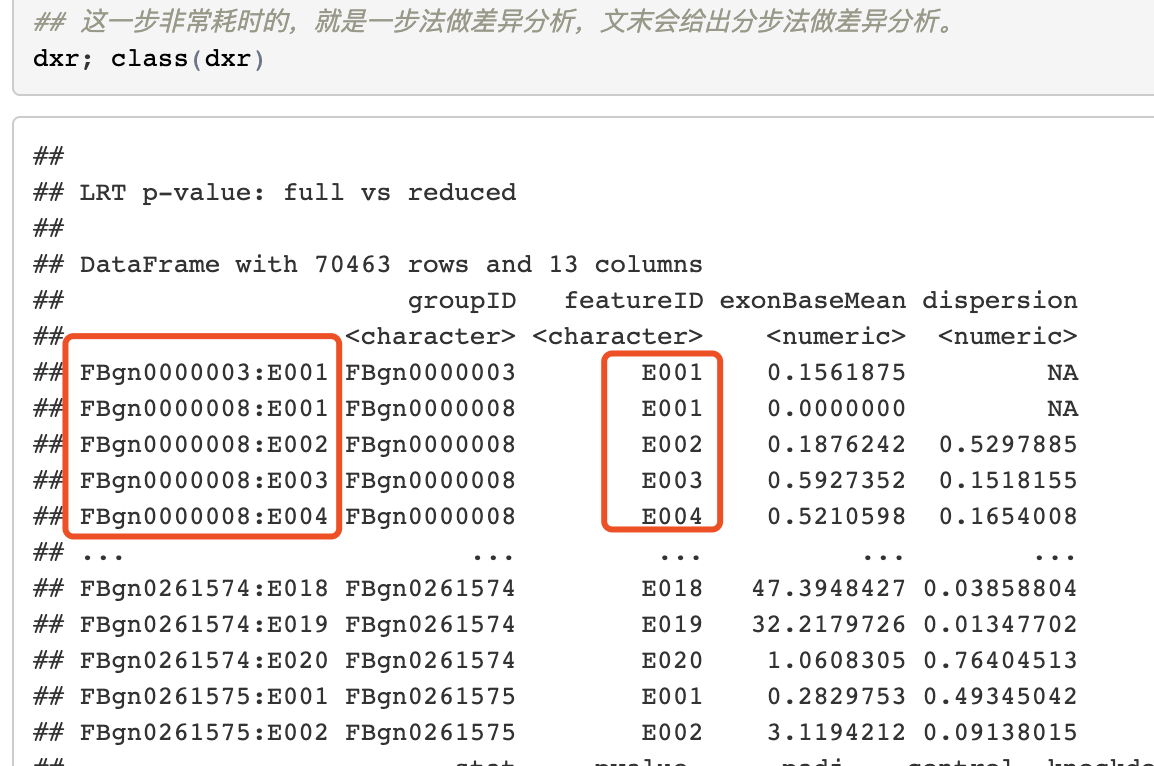
官网提供的是HTseq-count软件的结果转为DEXSeq输入的脚本,因为HTseq-count软件速度实在是不敢恭维,我还是想使用featurecounts。虽然它并没有DEXSeq输入的接口,但是简单谷歌,就可以搜得到解决方法;https://github.com/vivekbhr/Subread_to_DEXSeq
其实就是因为它需要不一样的gtf文件和gff文件。
安装GitHub插件处理gencode数据库的gtf文件
这里采用最原始的下载即可: https://github.com/vivekbhr/Subread_to_DEXSeq.git
cd
cd biosoft
git clone https://github.com/vivekbhr/Subread_to_DEXSeq.git
cd Subread_to_DEXSeq
wget ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_32/gencode.v32.annotation.gtf.gz
gunzip gencode.v32.annotation.gtf.gz
使用GitHub代码对gtf文件进行预处理:
which python
which pip
pip install HTSeq
cd ~/biosoft/Subread_to_DEXSeq
python dexseq_prepare_annotation2.py -f out_gv32.gtf gencode.v32.annotation.gtf out_gv32.gff
速度很快,得到的文件如下:
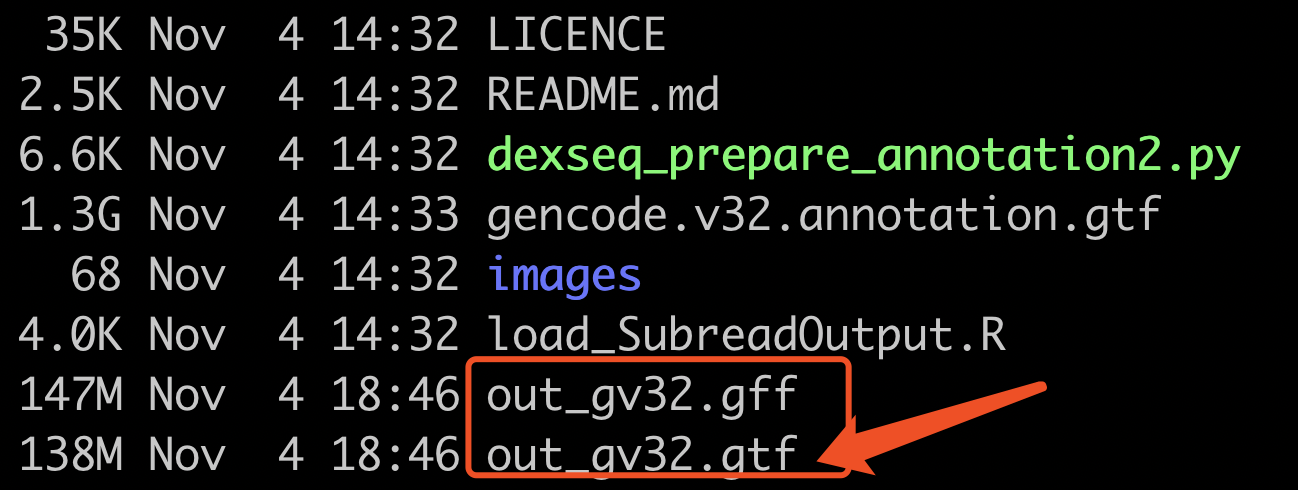
这个时候,需要记住的是,gtf文件可以被featureCounts用来定量,而后缀为gff的文件,是DEXSeq包需要的。
检查两个文件
首先看看后缀为gff的文件,是DEXSeq包需要的:
suppressPackageStartupMessages(library("DEXSeq"))
suppressPackageStartupMessages(library("pasilla"))
inDir <- system.file("extdata", package="pasilla")
gffFile <- list.files(inDir, pattern="gff$", full.names=TRUE)
gffFile
我的电脑的DEXSeq包举例的这个gff文件 在:
/Library/Frameworks/R.framework/Versions/3.5/Resources/library/pasilla/extdata/Dmel.BDGP5.25.62.DEXSeq.chr.gff
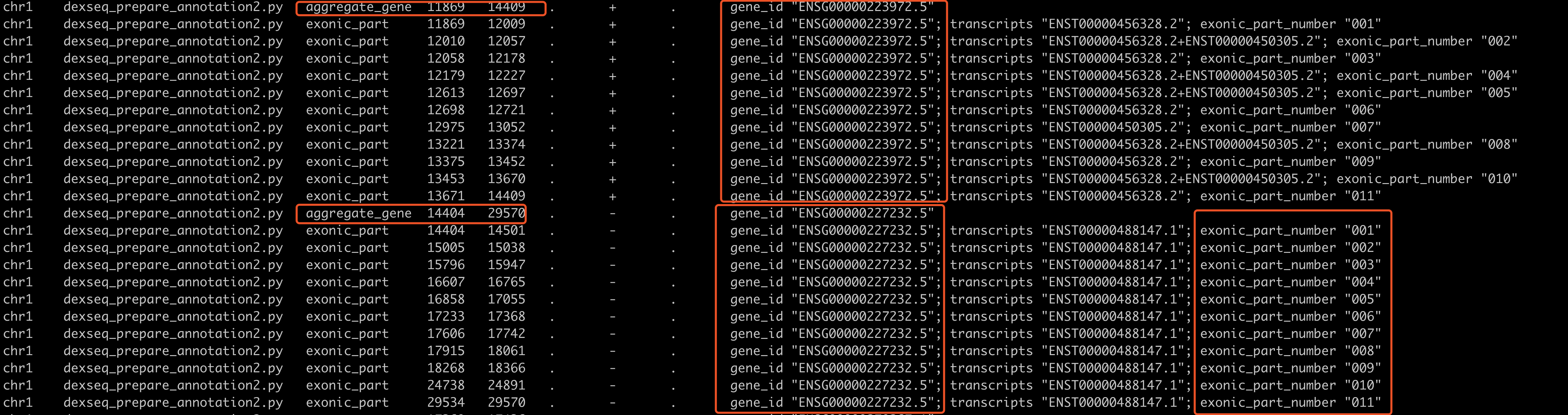
内容如下所示
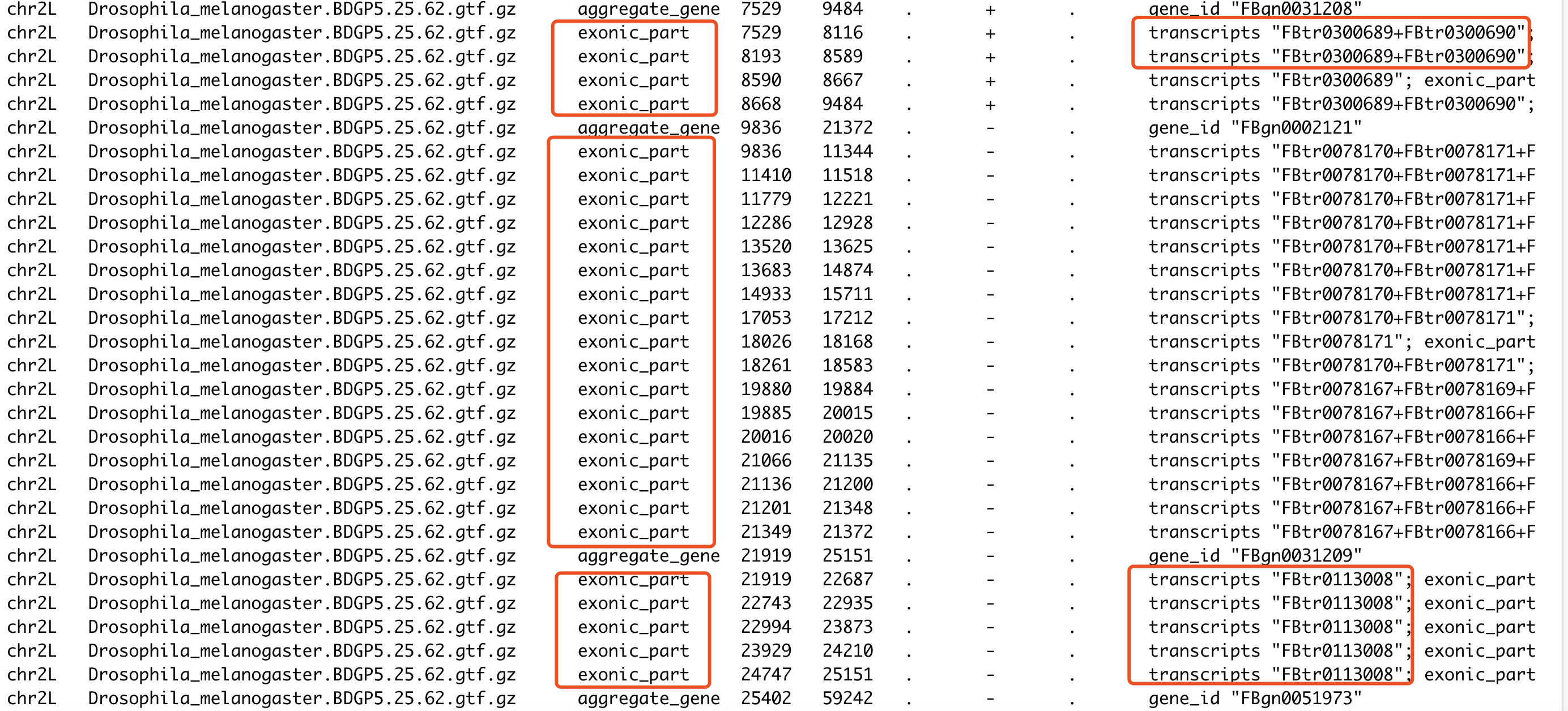
可以看到我们针对gencode数据库的gtf文件的处理,得到的文件也是符合要求的,跟这个R包自带的果蝇的例子类似,就是记录每个基因的多个转录本坐标,一个基因有多个转录本,就融合一下。
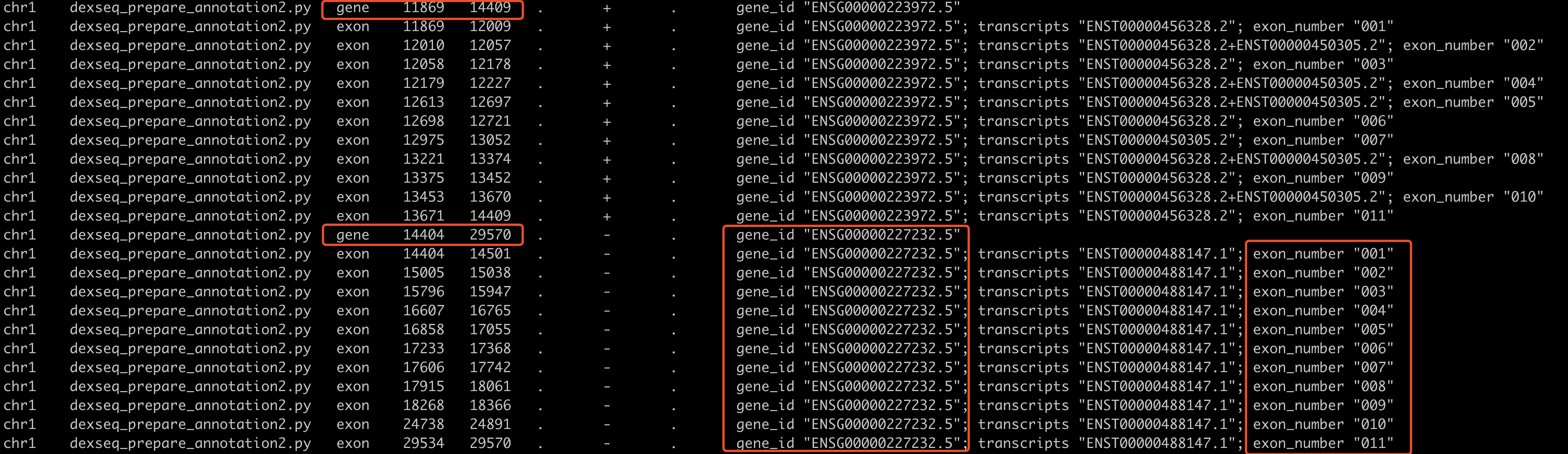
然后看看gtf文件,可以看到跟gff文件的区别几乎没有。
使用featureCounts定量
接下来就可以使用featureCounts对我们的bam文件进行定量啦,先看看示例数据:
suppressPackageStartupMessages(library("DEXSeq"))
suppressPackageStartupMessages(library("pasilla"))
inDir <- system.file("extdata", package="pasilla")
countFiles <- list.files(inDir, pattern="fb.txt$", full.names=TRUE)
countFiles
## 共七个样本,可以从文件名看到样本描述
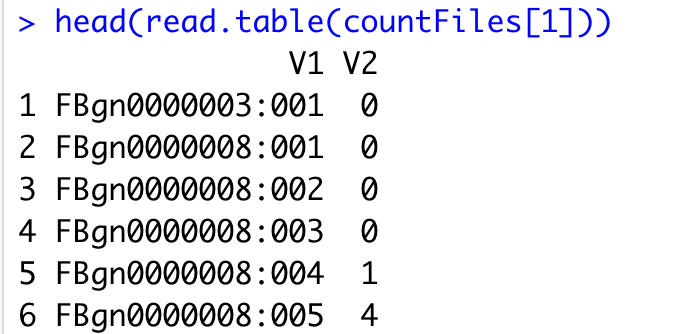
head(read.table(countFiles[1]))
下面的截图是HTseq-count软件的结果,
我们嫌弃HTseq-count软件运行速度太慢,所以才想着featureCounts,所以一切都得使用https://github.com/vivekbhr/Subread_to_DEXSeq 的脚本了。
conda activate rna
gtf=/home/yb77613/biosoft/Subread_to_DEXSeq/gencode.v32.annotation.gtf
#gtf=/home/yb77613/biosoft/Subread_to_DEXSeq/out_gv32.gtf
featureCounts -f -O -p -T 4 -F GTF \
-a ok.gtf \
-o test.txt ../star/SRR2016941.bam
-o npc2_fCount.out.txt ../star/*bam 1>counts.id.log 2>&1 &
重点是 -O 参数, 统计所有重叠的的meta-features的reads (或-f参数指定的feature)。