昨天发布了 GEO数据库中国区镜像横空出世,粉丝们都很happy,因为确实解决了他们的一个拦路虎,以后下载GEO数据再也不用去网吧了。但是部分粉丝提出了更过分的要求,说自己没有服务器,我以前的教程:(重磅!价值一千元的R代码送给你)芯片探针序列的基因组注释 他们跟随起来很困难,希望我随便把所有的gpl也注释一波提供给大家。
这个,当然没有问题,就是需要时间来实现,主要是因为lncRNA芯片的探针设计的时候并不是依据基因组设计,而是mRNA和lncRNA本身序列设计的,所以探针是会跨越外显子的,这一点在官网问答也说的很清楚:
For the coding mRNA on the array, is it just the exon regions or is it the whole gene locus (introns and exons)? 参考:https://www.arraystar.com/service/faq/
For each protein-coding mRNA, we designed a probe targeting their specific exon or exon-junction regions.
假如我们采取基因组比对策略,会有一个bugs出现,我给大家实例演示一下bowtie和hisat的区别,反正都是Johns Hopkins University的科研人员开发的。bowtie和hisat的软件安装,数据库参考文件,索引构建等等准备工作这里就不赘述了。
首先使用bowtie1比对全部的fasta序列探针
下面是一个例子,我首先去下载制作了 GPL15314_seq2fa.fasta 文件,然后使用bowtie1比对,参数选择的解释也在下面:
bowtie1=/trainee/jmzeng/tools/bowtie-1.1.2/bowtie
# http://bowtie-bio.sourceforge.net/manual.shtml
# In -v mode, alignments may have no more than V mismatches, where V may be a number from 0 through 3 set using the -v option.
# -m, Suppress all alignments for a particular read or pair if more than <int> reportable alignments exist for it.
bowtie2=/trainee/jmzeng/tools/bowtie2-2.3.5.1/bowtie
fasta=/trainee/jmzeng/Probe_seqfasta/lncRNA/human/GPL15314_seq2fa.fasta
sample=GPL15314
# # GPL15314 Arraystar Human LncRNA microarray V2.0 (Agilent_033010 Probe Name version)
index=/trainee/jmzeng/genome_index/human/human
$bowtie1 -v 0 -m 1 -p 6 $index -f $fasta -S ${sample}.sam
得到的log日志是:
# reads processed: 60699
# reads with at least one reported alignment: 49162 (80.99%)
# reads that failed to align: 9043 (14.90%)
# reads with alignments suppressed due to -m: 2494 (4.11%)
Reported 49162 alignments to 1 output stream(s)
发现比对率有点低,然后我搜索了其中几个探针,去blat看看为什么比对不上,发现果然是外显子问题。我们的这个探针序列是60个碱基,使用bowtie1比对失败,就是因为它没办法把这个探针序列的60个碱基拆分成为两个部分,分开比对在参考基因组的不同区域。
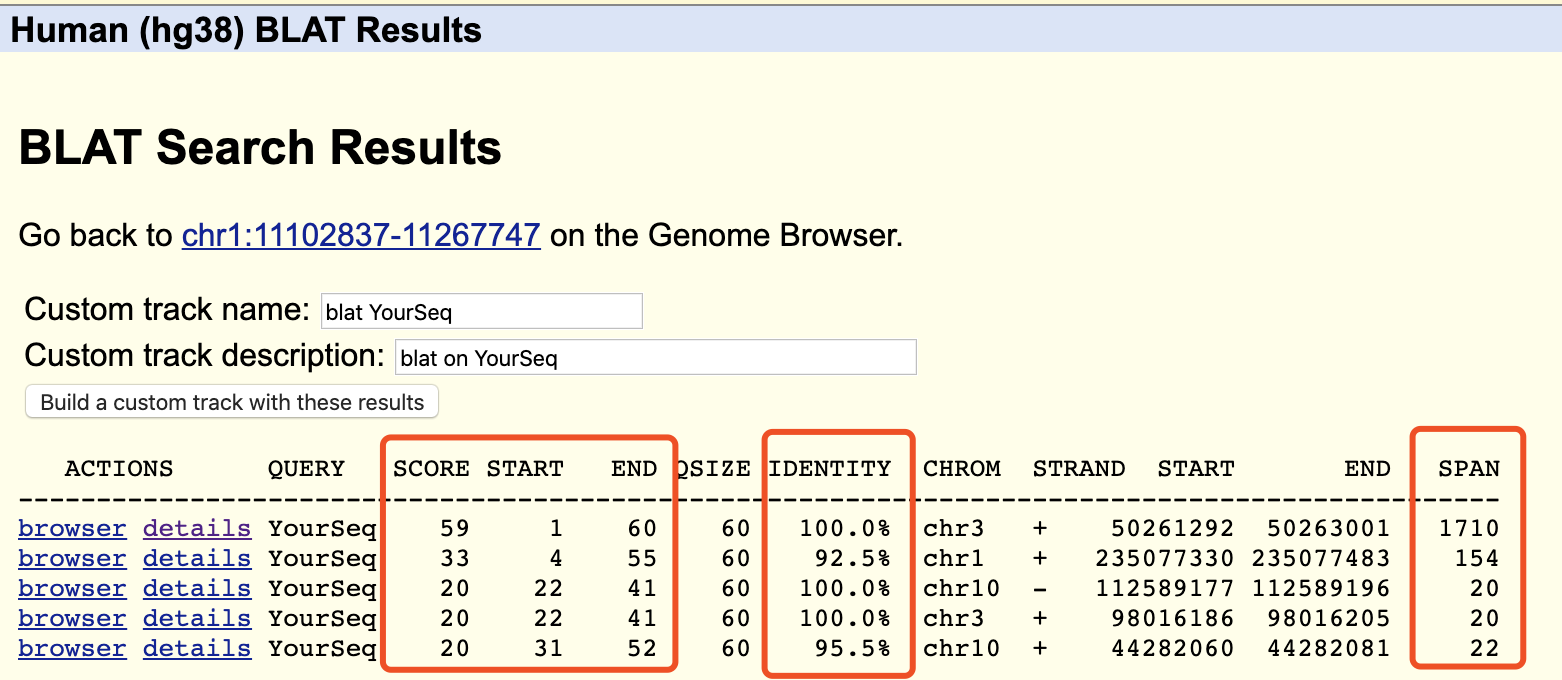
然后点击进入详情,可以看到我们的这个探针序列的60个碱基被拆分成为两个部分,分开比对在参考基因组的不同区域。
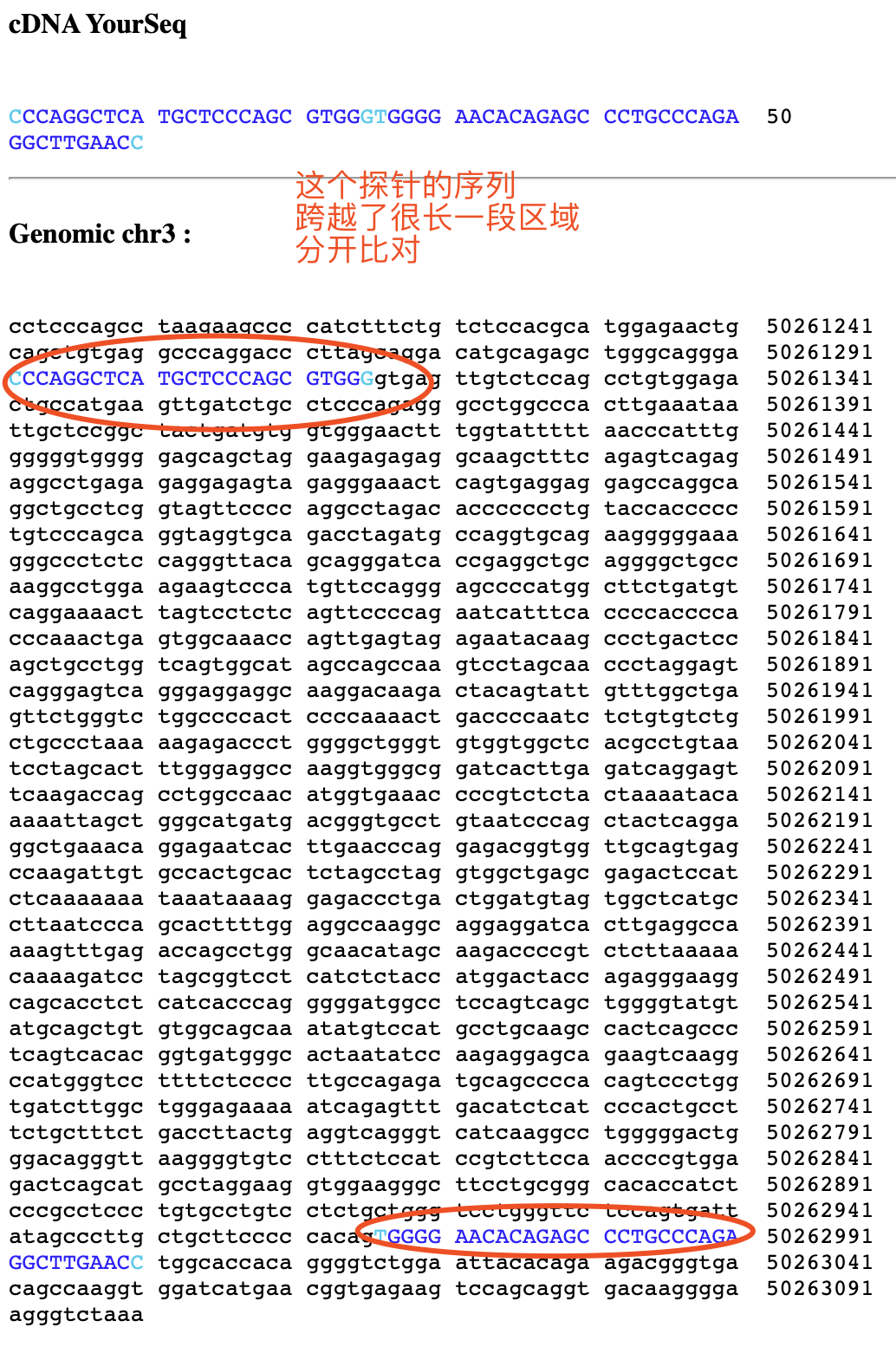
然后看看hisat
所以我们换一个比对工具,因为是需要跨越内含子的比对,所以选择hisat
hisat2=/trainee/jmzeng/tools//hisat2-2.0.0-beta/hisat2
fasta=/trainee/jmzeng/Probe_seqfasta/lncRNA/human/GPL15314_seq2fa.fasta
sample=GPL15314
index=/teach/database/index/hisat/hg38/genome
$hisat2 -f $fasta -x $index -S ${sample}.sam
然后发现
60699 reads; of these:
60699 (100.00%) were unpaired; of these:
519 (0.86%) aligned 0 times
54578 (89.92%) aligned exactly 1 time
5602 (9.23%) aligned >1 times
99.14% overall alignment rate
比对率不得了啊!
题外话
我很喜欢blat这个在线网页工具,因为当初听说它的速度甩blast工具几十条街。
在我B站视频,多次提到它的奇妙用法,但我也是今天才知道,它居然也可以跨越内含子进行比对。