前面我们系统性的总结了circRNA的相关背景知识:
同样的策略,我们也可以应用到lncRNA的学习。所以昨天我们发布了:lncRNA的一些基础知识 ,那么接下来我们需要分享的就是lncRNA芯片的一般分析流程和lncRNA-seq数据的一般分析流程!
GEO数据库里面的lncRNA芯片平台很多
与circRNA芯片不同的是,GEO数据库里面的lncRNA芯片平台很多,毕竟已经是三五年前的热点了。
如果按照使用量排序,可以看到其实也就几百个数据集而已。
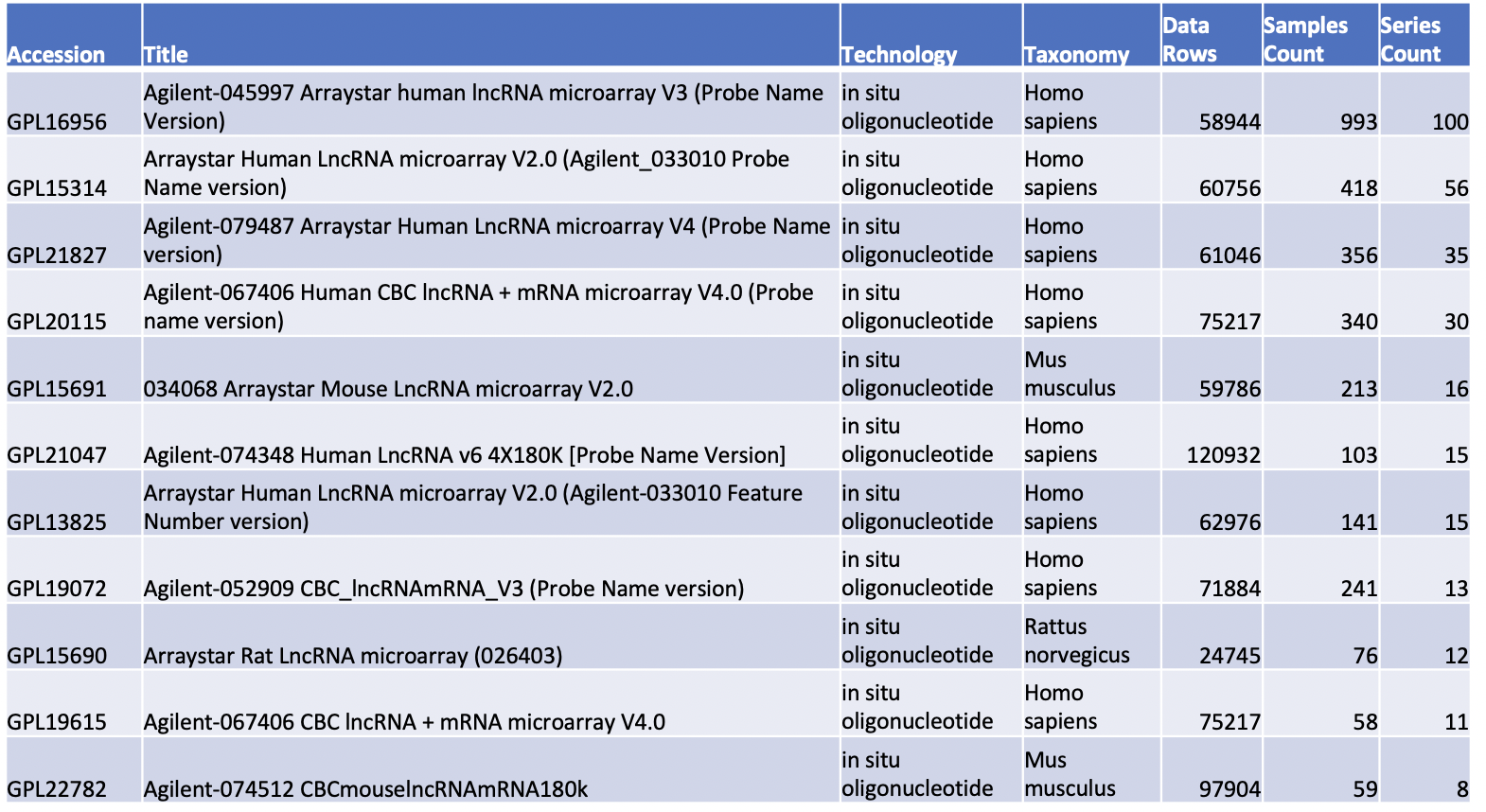
感兴趣的朋友可以点击进入查看详情。每个芯片平台的主页内容都是非常丰富的。
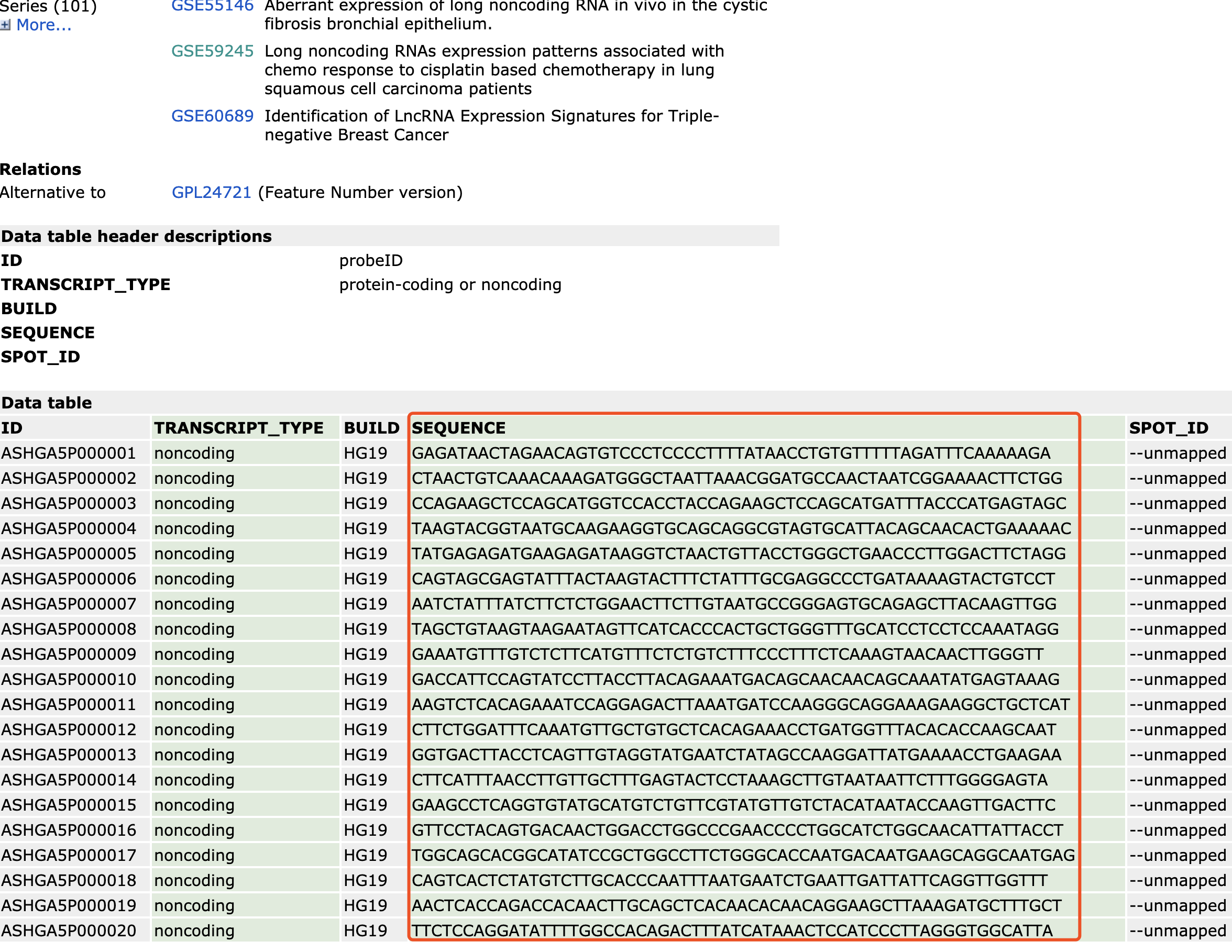
通常lncRNA芯片只有探针序列
比如:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL16956 ,如下所示,有一百多个数据集都是使用的这个平台的芯片,但是可以看到,其GEO数据库主页里面,仅仅含有每个探针的碱基序列,并没有更丰富的lncRNA基因注释信息。
进入其中一个实例:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE59245 我们可以学习一下,如何使用这样的lncRNA芯片。都是走标准分析流程,火山图,热图,GO/KEGG数据库注释等等。这些流程的视频教程都在B站和GitHub了,目录如下:
- 第一讲:GEO,表达芯片与R
- 第二讲:从GEO下载数据得到表达量矩阵
- 第三讲:对表达量矩阵用GSEA软件做分析
- 第四讲:根据分组信息做差异分析
- 第五讲:对差异基因结果做GO/KEGG超几何分布检验富集分析
- 第六讲:指定基因分组boxplot指定基因list画热图
仅仅是最后得到的差异分子,并不是以前的mRNA后面的基因名,而是miRNA,lncRNA,甚至circRNA的ID,看起来很陌生罢了。感兴趣可以细读表达芯片的公共数据库挖掘系列推文 ;
- 解读GEO数据存放规律及下载,一文就够
- 解读SRA数据库规律一文就够
- 从GEO数据库下载得到表达矩阵 一文就够
- GSEA分析一文就够(单机版+R语言版)
- 根据分组信息做差异分析- 这个一文不够的
- 差异分析得到的结果注释一文就够
常规的表达量分析实验设计
来一个示例吧,这篇文章(Long noncoding RNAs expression patterns associated with chemo response to cisplatin based chemotherapy in lung squamous cell carcinoma patients. PLoS One 2014; PMID: 25250788) 使用的是 GPL16956 Agilent-045997 Arraystar human lncRNA microarray V3 (Probe Name Version)
分成2个组,然后走差异分析流程即可:
GSM1431249 69_PR
GSM1431250 80_PR
GSM1431251 83_PR
GSM1431252 141_PR
GSM1431253 164_PR
GSM1431254 165_PD
GSM1431255 169_PD
GSM1431256 175_PD
GSM1431257 177_PD
GSM1431258 198_PD
主要的结论就是根据什么阈值挑选到了多个上下调的lncRNAs基因:
Compared with the PD samples, 953 lncRNAs were consistently upregulated and 749 lncRNAs were downregulated consistently among the differentially expressed lncRNAs in PR samples (Fold Change≥2.0-fold, p <0.05).
因为是探索的是 lung squamous cell carcinoma 病人的chemo response to cisplatin,所以这个差异分析结果也正好跟”Nucleotide excision repair,” 等生物学功能富集。学徒作业:下载这个文章的这个数据集也走差异分析流程,并且进行GO/KEGG数据库功能富集注释看看是否与文章相符合。
芯片里面是有 lncRNA 和 mRNA
就可以分开分析,分开走差异流程,分开富集分析,这样的话图表就多一点。比如文章就展示了两个热图,如下:
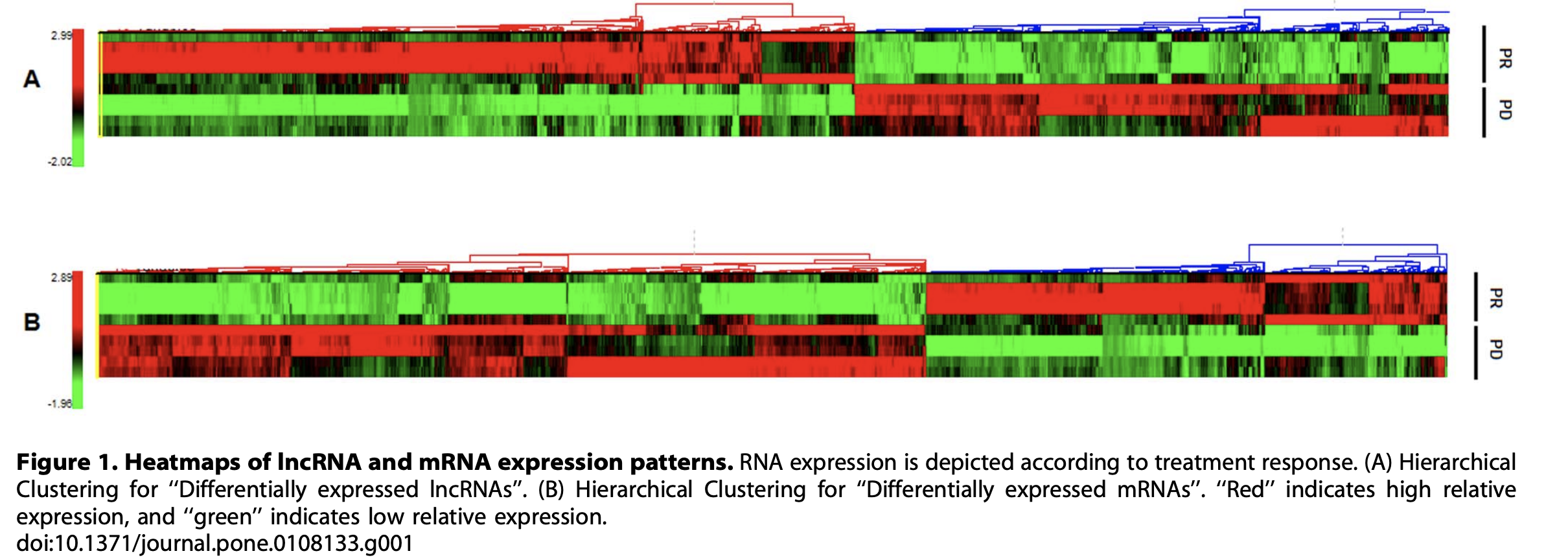
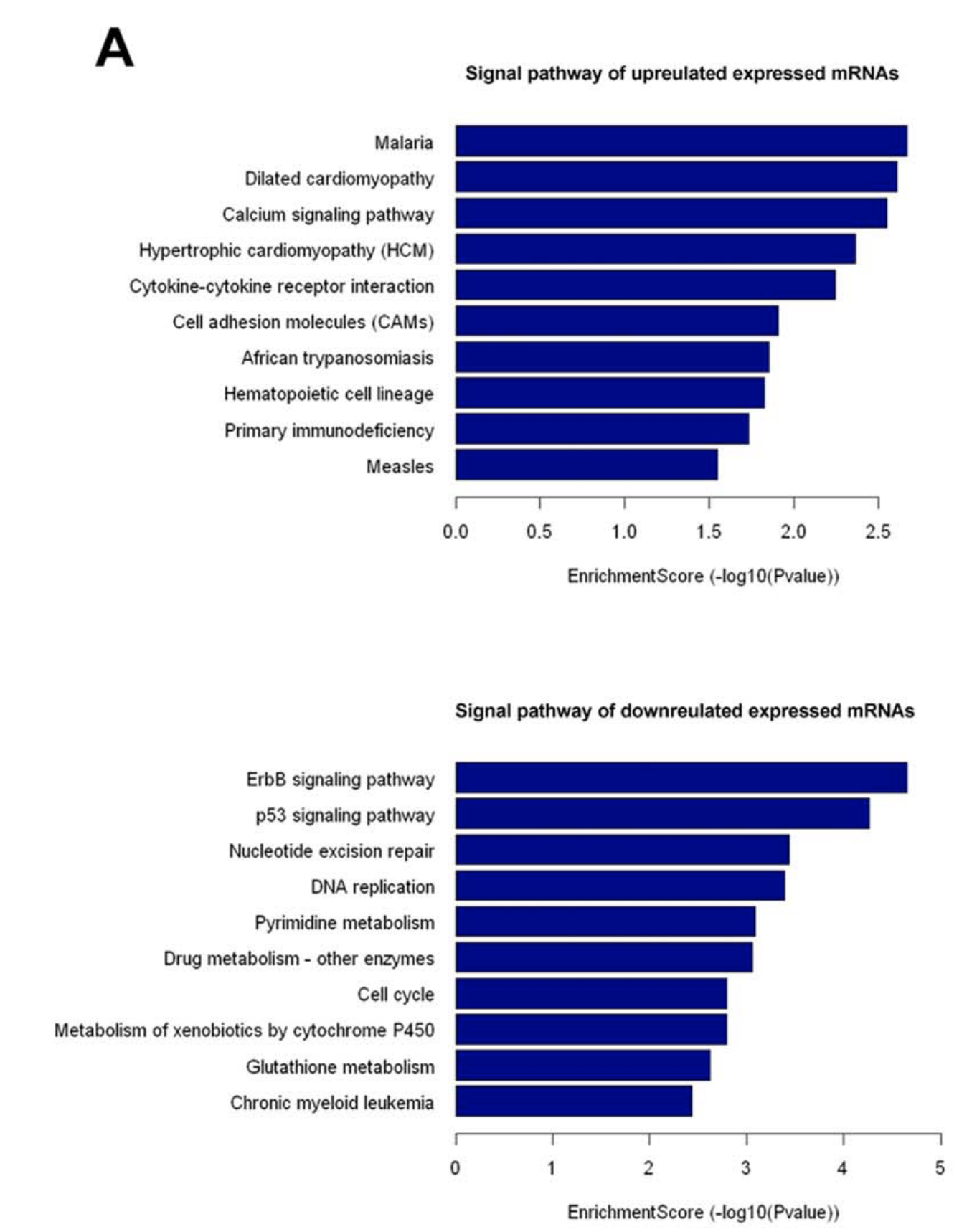
富集分析
可以看到,作者这里是把编码mRNA的基因分成统计学显著的上下调后,然后分开做KEGG数据库的超几何分布检验啦。可以看到,下调基因里面的功能排在第三的就是”Nucleotide excision repair,”符合作者的实验设计,肺癌的cisplatin耐药。
lncRNA芯片数据结果可以只是一个引子
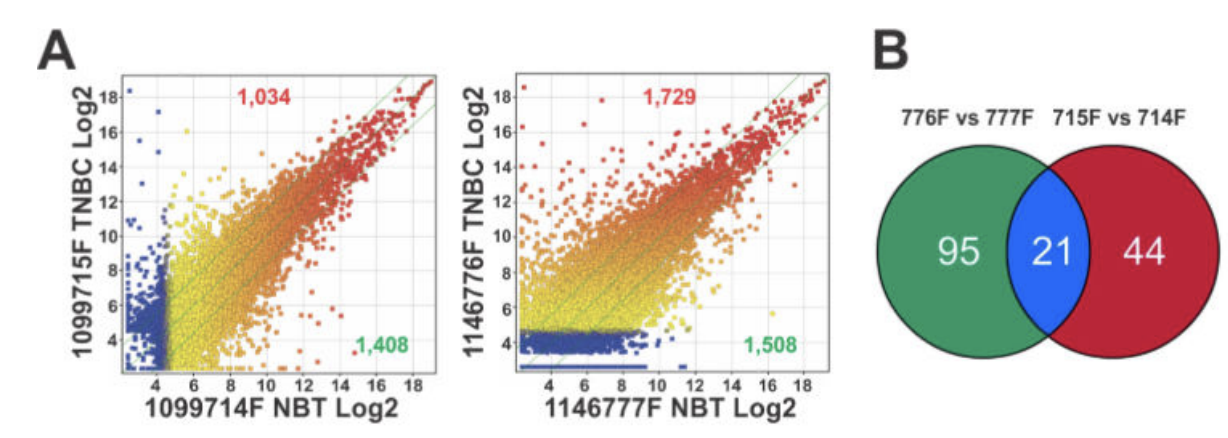
比如 GSE60689 Identification of LncRNA Expression Signatures for Triple-negative Breast Cancer , 文章是 lncRNA directs cooperative epigenetic regulation downstream of chemokine signals. Cell 2014 Nov PMID: 25416949 , 仅仅是4个样本,两个分组。
GSM1485226 Normal adjacent breast tissue [73]
GSM1485227 Infiltrating Ductal Carcinima of breast [73]
GSM1485228 Infiltrating Ductal Carcinima of breast [67]
GSM1485229 Normal adjacent breast tissue [67]
但是该做的分析一点都不会少,仍然是 Scatter plots of lncRNAs significantly up-regulated (red) or down-regulated (green) in two pairs of TNBC tissues compared to the matched adjacent normal tissues (NBT).
有趣的是,相当于这个文章的芯片其实是并没有生物学重复,两个病人独立的分析,然后取两个病人的差异基因的交集作为后续实验验证,因为这个cell文章,表达芯片的差异分析结果只是人家生物学故事的一个引子。
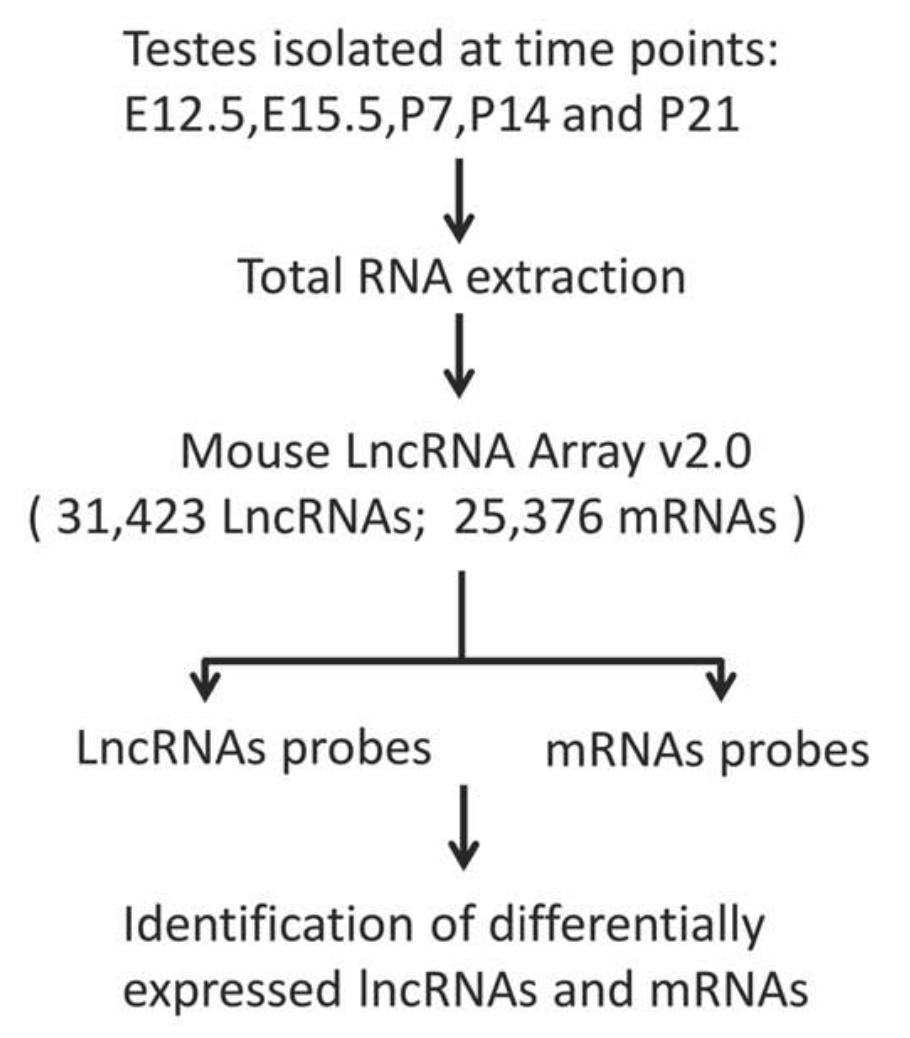
在小鼠的lncRNA芯片数据集例子
看数据集:GSE46896,其文章发表在一个很普通的杂志:Biol Reprod. 2013 Nov 7; 标题是:Expression profiling reveals developmentally regulated lncRNA repertoire in the mouse male germline 实验设计的很好,主要是分析太简陋了,生物学故事很一般。而且全文绘图都是Excel表格样式:
因为其分组比较多,所以可分析的点也会相应的多。
其实主要是共调控网络分析
共调控网络分析其实是公共数据库挖掘的一个很大众的方向,只要有两个表达矩阵,就可以分开独立走差异分析得到基因集,后续通过数据库进行关联,从而构建网络。
比如文章 Non-coding RNAs participate in the regulatory network of CLDN4 via ceRNA mediated miRNA evasion. Nat Commun 2017 Aug PMID: 28819095
GSM2643709 non-tumorous adjacent tissues_rep1
GSM2643710 non-tumorous adjacent tissues_rep2
GSM2643711 non-tumorous adjacent tissues_rep3
GSM2643712 non-tumorous adjacent tissues_rep4
GSM2643713 non-tumorous adjacent tissues_rep5
GSM2643714 non-tumorous adjacent tissues_rep6
GSM2643715 Gastric cancer tissues_rep1
GSM2643716 Gastric cancer tissues_rep2
GSM2643717 Gastric cancer tissues_rep3
GSM2643718 Gastric cancer tissues_rep4
GSM2643719 Gastric cancer tissues_rep5
GSM2643720 Gastric cancer tissues_rep6
就是分不同类型( lncRNA 和 mRNA )的基因,走差异分析流程,多个火山图,多个热图,最后挑选统计学显著的差异基因构建网络。所以需要加上miRNA芯片。
In an attempt to identify the regulatory networks of mRNA and ncRNAs in GC, six pairs of GC tissues and non-tumorous adjacent tissues were analyzed via microarray using the Human LncRNA+mRNA Array v3.0 together with the miRCURY LNATM microRNA Array.
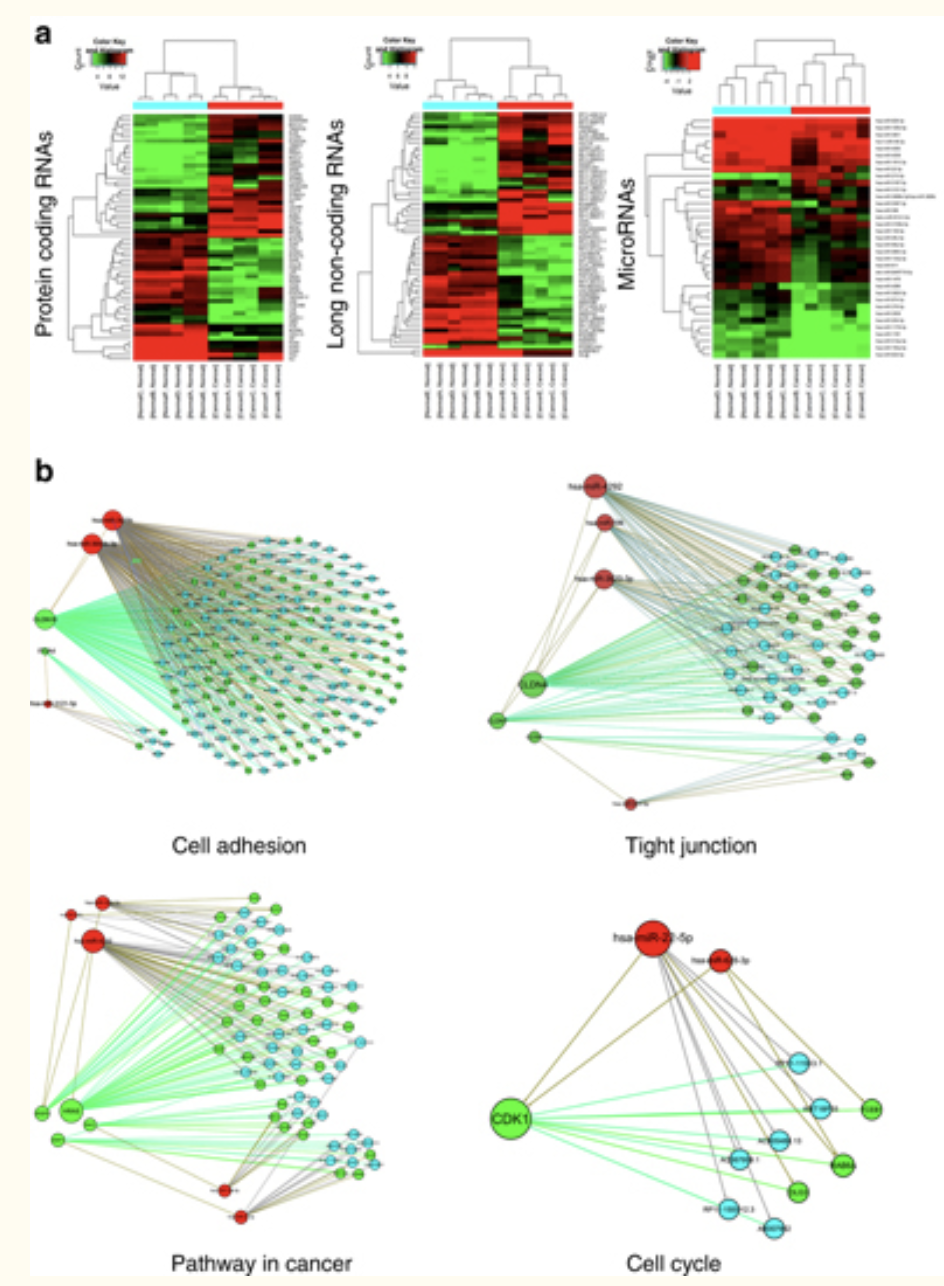
类似的LncRNA + mRNA array 研究非常多,再比如文章:Long noncoding RNA expression profile reveals lncRNAs signature associated with extracellular matrix degradation in kashin-beck disease
LncRNA and mRNA expression profiling were performed using Agilent human lncRNA + mRNA array 4.0 platform (4 × 180 K), with each array containing approximately 41,000 lncRNA and 34,000 mRNA probes.
In this study, we identified 316 up-regulated and 631 down-regulated lncRNAs (≥ 2-fold change) in KBD chondrocytes
We also identified 232 up-regulated and 427 down-regulated mRNAs (≥ 2-fold change).
然后就针对具有统计学显著表达差异的LncRNA + mRNA 构建网络。
一个简单的例子
希望你安装一下AnnoProbe包,然后走下面的代码。
rm(list = ls())
library(AnnoProbe)
library(ggpubr)
suppressPackageStartupMessages(library(GEOquery))
getwd()
setwd('test/')
## download GSE95186 data
# https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE95186
# eSet=getGEO('GSE95186', destdir=".", AnnotGPL = F, getGPL = F)[[1]]
# 使用 GEO 中国区镜像进行加速
gset=AnnoProbe::geoChina('GSE95186')
gset
# check the ExpressionSet
eSet=gset[[1]]
probes_expr <- exprs(eSet);dim(probes_expr)
head(probes_expr[,1:4])
boxplot(probes_expr,las=2)
## pheno info
phenoDat <- pData(eSet)
head(phenoDat[,1:4])
# https://www.ncbi.nlm.nih.gov/pubmed/31430288
group_list=factor(c(rep('treat',3),rep('untreat',3)))
table(group_list)
eSet@annotation
# GPL15314 Arraystar Human LncRNA microarray V2.0 (Agilent_033010 Probe Name version)
GPL=eSet@annotation
# 选择 pipe 获取的是 冗余注释,也就是说一个探针很有可能会对应多个基因。
probes_anno <- idmap(GPL,type = 'pipe')
head(probes_anno)
probes_anno=probes_anno[probes_anno$probe_id %in% rownames(probes_expr),]
# 只需要表达矩阵里面有的探针的注释即可
length(unique(probes_anno$probe_id))
anno=annoGene(probes_anno$symbol,'SYMBOL')
head(anno)
pcs=probes_anno[probes_anno$symbol %in% anno[anno$biotypes=='protein_coding',1],]
nons=probes_anno[probes_anno$symbol %in% anno[anno$biotypes !='protein_coding',1],]
# 可以首先把探针拆分成为 protein_coding 与否
length(unique(pcs$probe_id))
length(unique(nons$probe_id))
# 可以看到仍然是有探针会被注释到多个基因,这个时候
pcs_expr <- probes_expr[pcs$probe_id,]
nons_expr <- probes_expr[nons$probe_id,]
boxplot(pcs_expr,las=2)
boxplot(nons_expr,las=2)
# 很容易看出来,非编码的这些基因的平均表达量,是低于编码的。
## 首先对编码基因的表达矩阵做差异分析
genes_expr <- filterEM(pcs_expr,pcs )
if(T){
head(genes_expr)
library("FactoMineR")
library("factoextra")
dat.pca <- PCA(t(genes_expr) , graph = FALSE)
dat.pca
fviz_pca_ind(dat.pca,
geom.ind = "point",
col.ind = group_list,
addEllipses = TRUE,
legend.title = "Groups"
)
library(limma)
design=model.matrix(~factor(group_list))
design
fit=lmFit(genes_expr,design)
fit=eBayes(fit)
DEG=topTable(fit,coef=2,n=Inf)
head(DEG)
## visualization
need_deg=data.frame(symbols=rownames(DEG), logFC=DEG$logFC, p=DEG$P.Value)
deg_volcano(need_deg,1)
deg_volcano(need_deg,2)
deg_heatmap(DEG,genes_expr,group_list)
deg_heatmap(DEG,genes_expr,group_list,30)
}
### 然后对非编码的基因的表达矩阵做差异分析
genes_expr <- filterEM(nons_expr,nons )
if(T){
head(genes_expr)
library("FactoMineR")
library("factoextra")
dat.pca <- PCA(t(genes_expr) , graph = FALSE)
dat.pca
fviz_pca_ind(dat.pca,
geom.ind = "point",
col.ind = group_list,
addEllipses = TRUE,
legend.title = "Groups"
)
library(limma)
design=model.matrix(~factor(group_list))
design
fit=lmFit(genes_expr,design)
fit=eBayes(fit)
DEG=topTable(fit,coef=2,n=Inf)
head(DEG)
## visualization
need_deg=data.frame(symbols=rownames(DEG), logFC=DEG$logFC, p=DEG$P.Value)
deg_volcano(need_deg,1)
deg_volcano(need_deg,2)
deg_heatmap(DEG,genes_expr,group_list)
deg_heatmap(DEG,genes_expr,group_list,30)
}
需要使用下面的代码自行下载安装我们的AnnoProbe包
library(devtools)
install_github("jmzeng1314/AnnoProbe")
library(AnnoProbe)
因为这个包里面并没有加入很多数据,所以理论上会比较容易安装,当然,不排除中国大陆少部分地方基本上连GitHub都无法访问。配合着详细的介绍:
因为这些包暂时托管在GitHub平台,但是非常多的朋友访问GitHub困难,尤其是我打包了好几百个GPL平台的注释信息后, 我的GitHub包变得非常臃肿,大家下载安装困难,所以我重新写一个精简包。也在:芯片探针ID的基因注释以前很麻烦 和 :芯片探针序列的基因注释已经无需你自己亲自做了, 里面详细介绍了。最重要的是idmap函数,安装方法说到过:芯片探针序列的基因注释已经无需你自己亲自做了, 也有交流群,可以反馈使用过程的体验。