我特别不喜欢写网页工具或者鼠标点点点的软件操作指南,因为感觉就跟QQ软件一样,自己摸索就ok了,所以我的cytoscape十讲就一直处于施工阶段:
- Cytoscape十讲之网络图的认知
- Cytoscape十讲之下载
每次都是写到一半,就弃稿了!
不过,好在我有一千多学员,一百多个学徒,给他们安排的作业就是写这些简单软件操作指南,这样就弥补了我写不来太基础教程的弱点。
不太长的总结版本,基因的蛋白互作网络图可以类比于人群社会关系连接图,然后子网络hub基因,类似于人群里面的不同派系,各个派系的首脑人物!
- Cytoscape十讲之下载
正文分割线,作者JOJO
刚刚接触cytoscape,应该怎么学习呢?感觉这个软件参数贼多,还是英文,一看就头大,怎么入手呢?JOJO表示不慌,下面是一些JOJO熟悉cytoscape的思路,可供参考。
首先,找个模板操作操作,就是软件安装后的自带的示例数据:
模板探索:
1.打开cytoscape,看到sample sessions,一看箭头所指网络图可以说非常符合直男审美了,打开打开打开
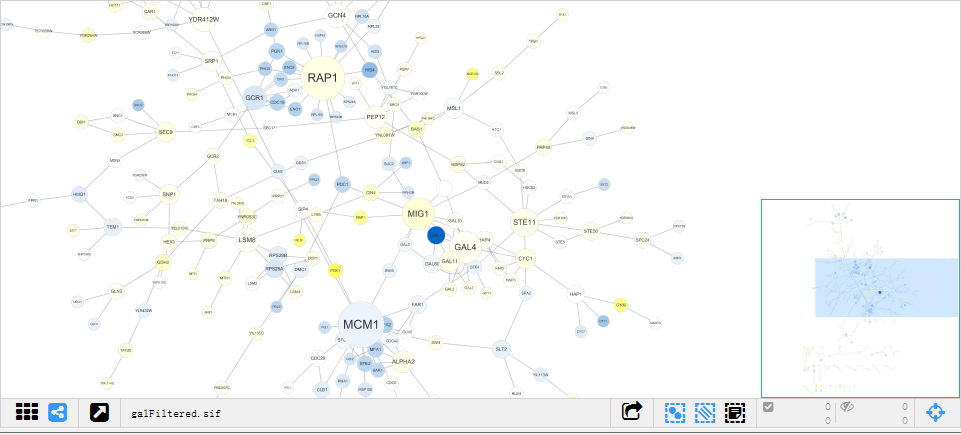
这样的一个图,感觉很吸引眼球。那么这样一个图是怎么出来的呢,要想知道它是什么,就去找生成这个图的数据,打开cytoscape文件中的sampledata可以看到以galfiltered命名的一些各种格式的文件(.sif/.nnf/.XGMML/.csv等)。关于这些不同的格式,网上有详细的讲解,JOJO对于这些的理解就是一批数据不同的载体,读入的过程也许有区别,但是它们都是服务于同一个网络图。
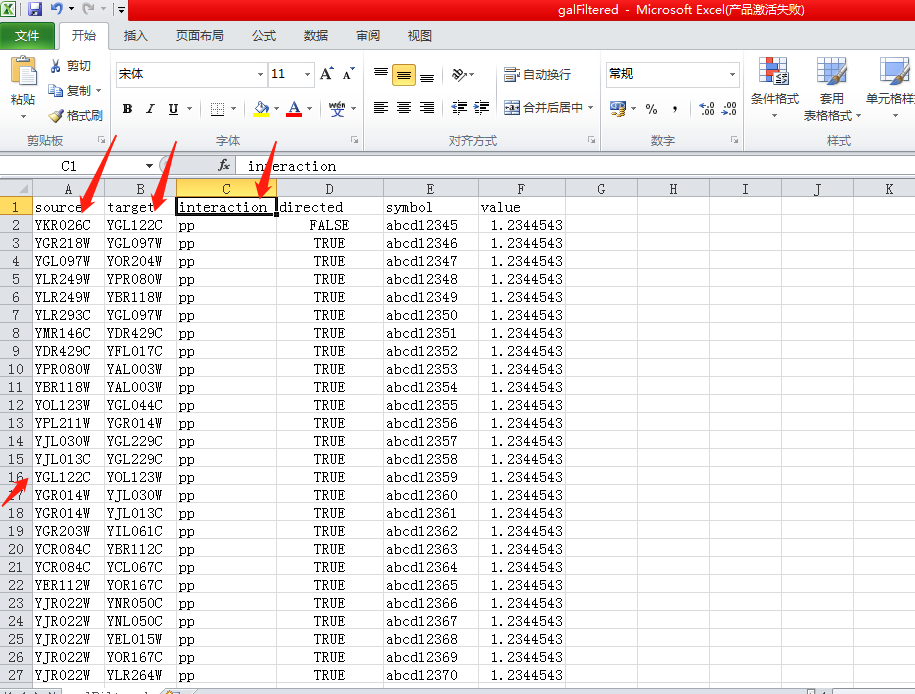
打开galfiltered.csv,可以看到source,target,interaction这样的3列,这就是source node,target node,interaction,对应网络图中的节点,及节点之间的互作关系,这里的interaction主要有两种pp(protein-protein),pd(protein-DNA)。这里有一个值得注意的点,一个基因可以既是source node又是target node,甚至可以同时是source node和target node(没见过不管)。如上图,YGL122C在第2、16行都有出现。
就好比杨过,他既是小龙女的侄子又是小龙女的丈夫
。
再看另一个galexpdata.csv文件,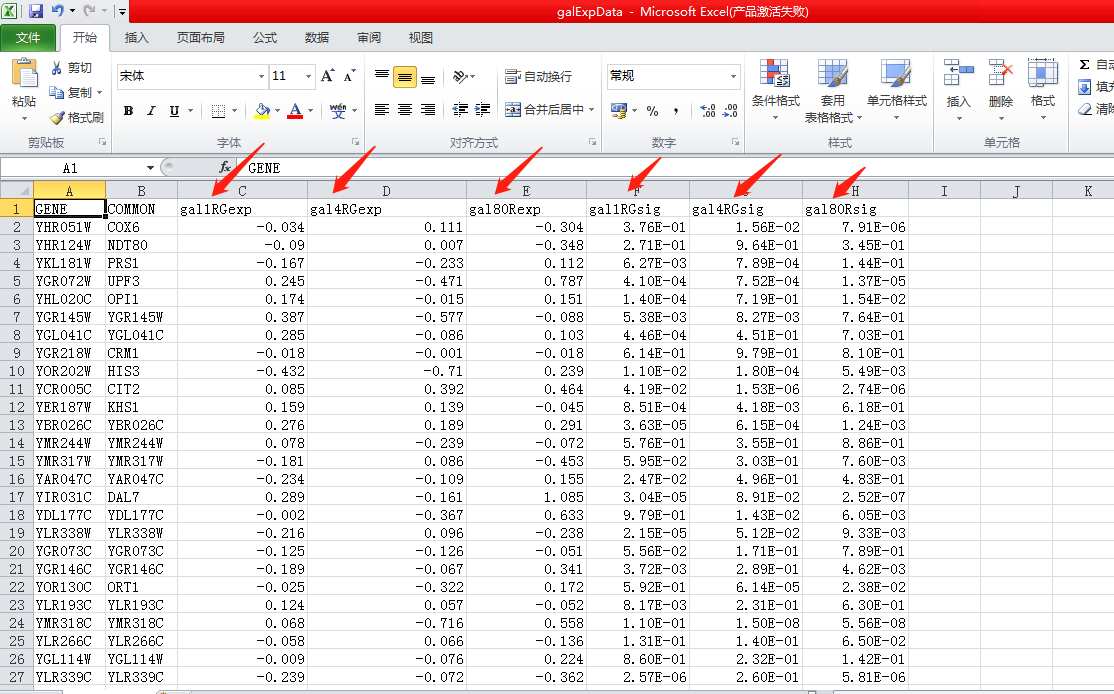 ,可以看出来这是一个属性文件,记录了基因的属性,也就是节点的各种属性,列名中进一步可以看到gal1,gal4,gal80,经查询后得知这是3种酵母转录因子,猜测每一行就是一个基因关gal1,gal4,gal80这3种酵母转录因子的表达量信息值,那么这3个转录因子必然在网络图中是关键节点,暂且记住,返回网络图再把它们找出来加深对于图的理解。那么对于数据的理解就暂且这样,下面导入数据。
,可以看出来这是一个属性文件,记录了基因的属性,也就是节点的各种属性,列名中进一步可以看到gal1,gal4,gal80,经查询后得知这是3种酵母转录因子,猜测每一行就是一个基因关gal1,gal4,gal80这3种酵母转录因子的表达量信息值,那么这3个转录因子必然在网络图中是关键节点,暂且记住,返回网络图再把它们找出来加深对于图的理解。那么对于数据的理解就暂且这样,下面导入数据。
2.分别使用这两个快捷键 ,将galfiltered.csv及galexpdata.csv导入。导入后如下:
,将galfiltered.csv及galexpdata.csv导入。导入后如下: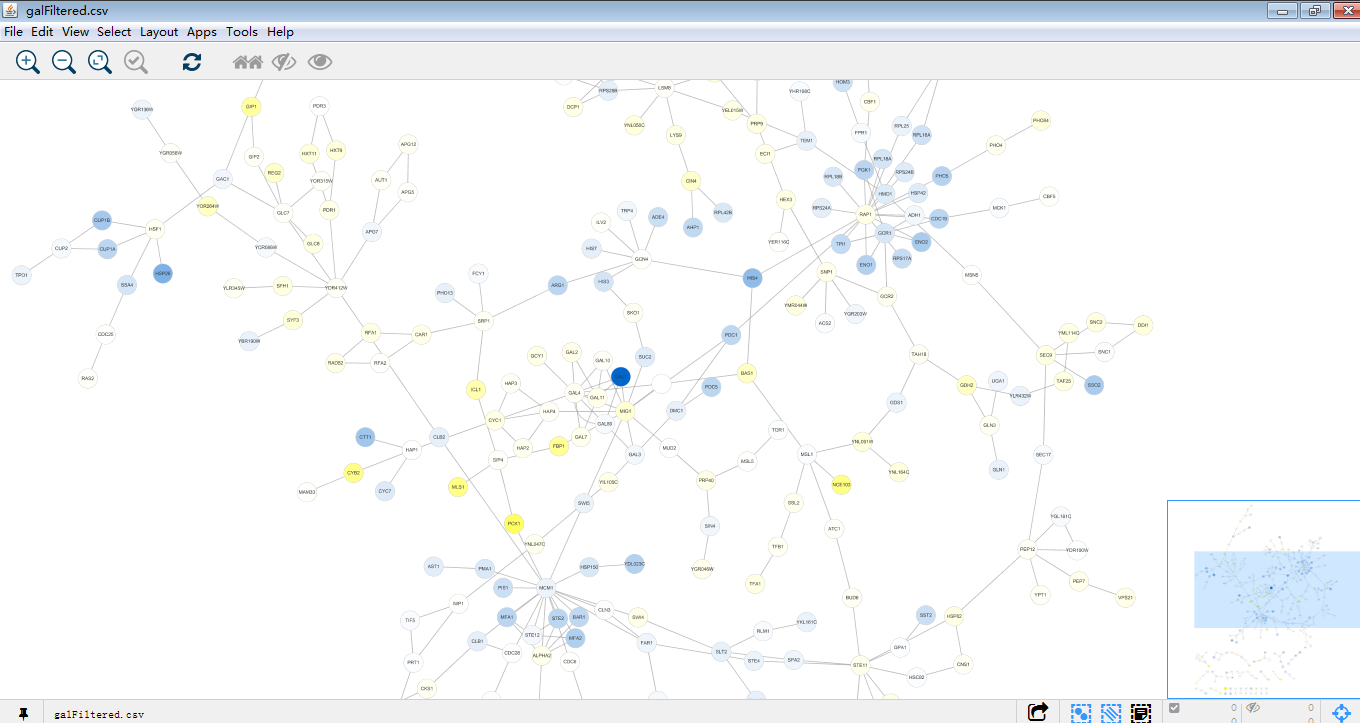
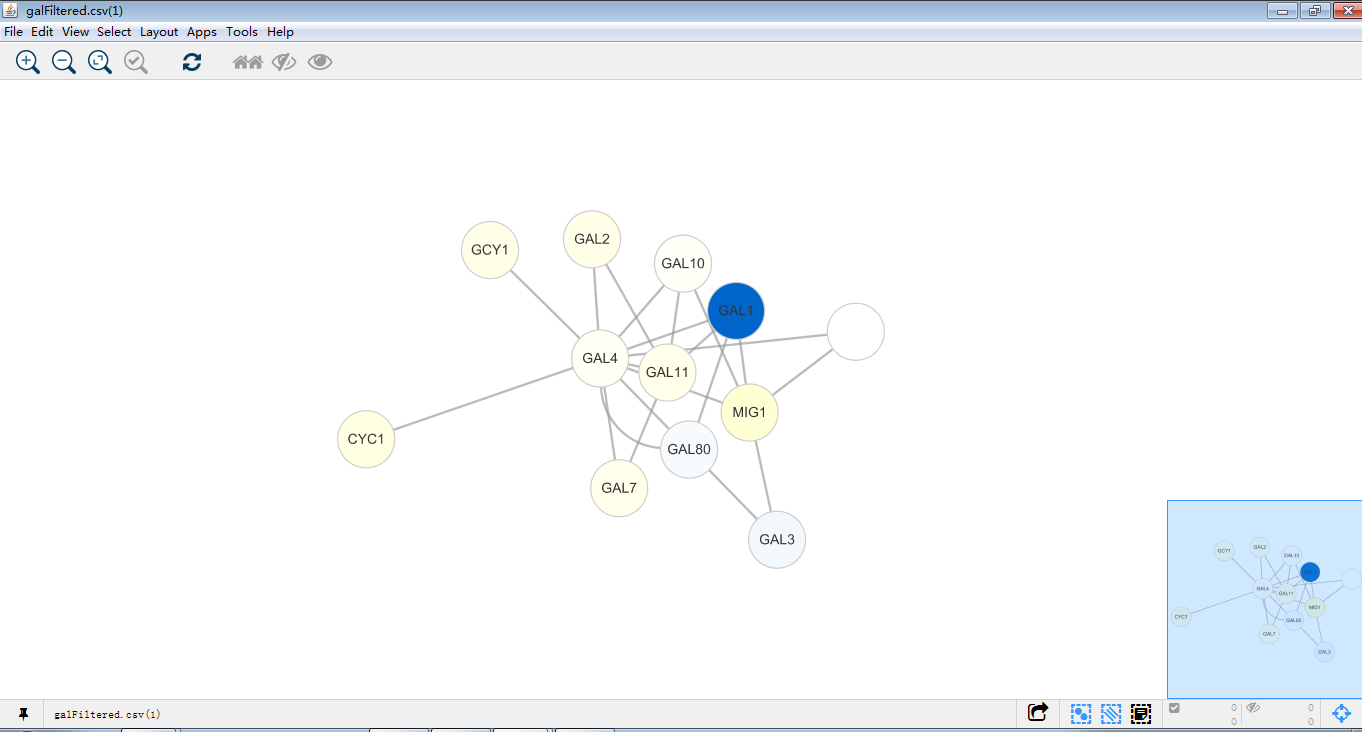
这就是一个漂亮的网络图了,再对这个图进行探索。首先找到gal1,gal4,gal80,在下方table panel里即为这个GAL1节点的详细信息,同时GAL1,GAL4,GAL80在图中位置一目了然。那么接下来试试将这一大家庭提出来。 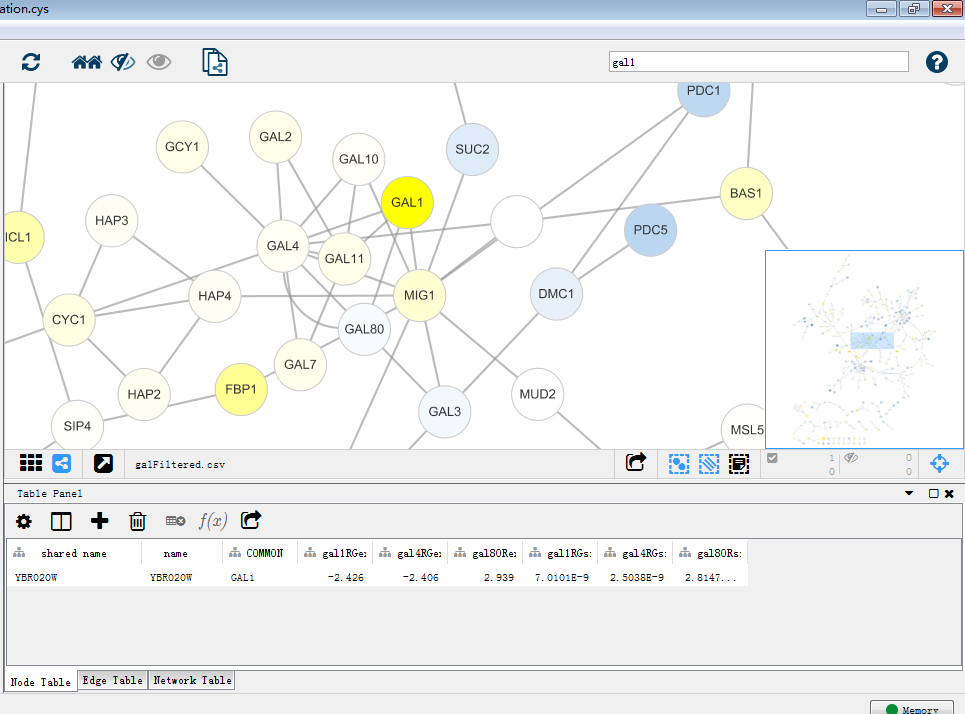
同时选中GAL1,GAL4,GAL80,依次点击箭头图标(当然也可以使用快捷键),即可将GAL1,GAL4,GAL80及邻近节点做一个新网络图。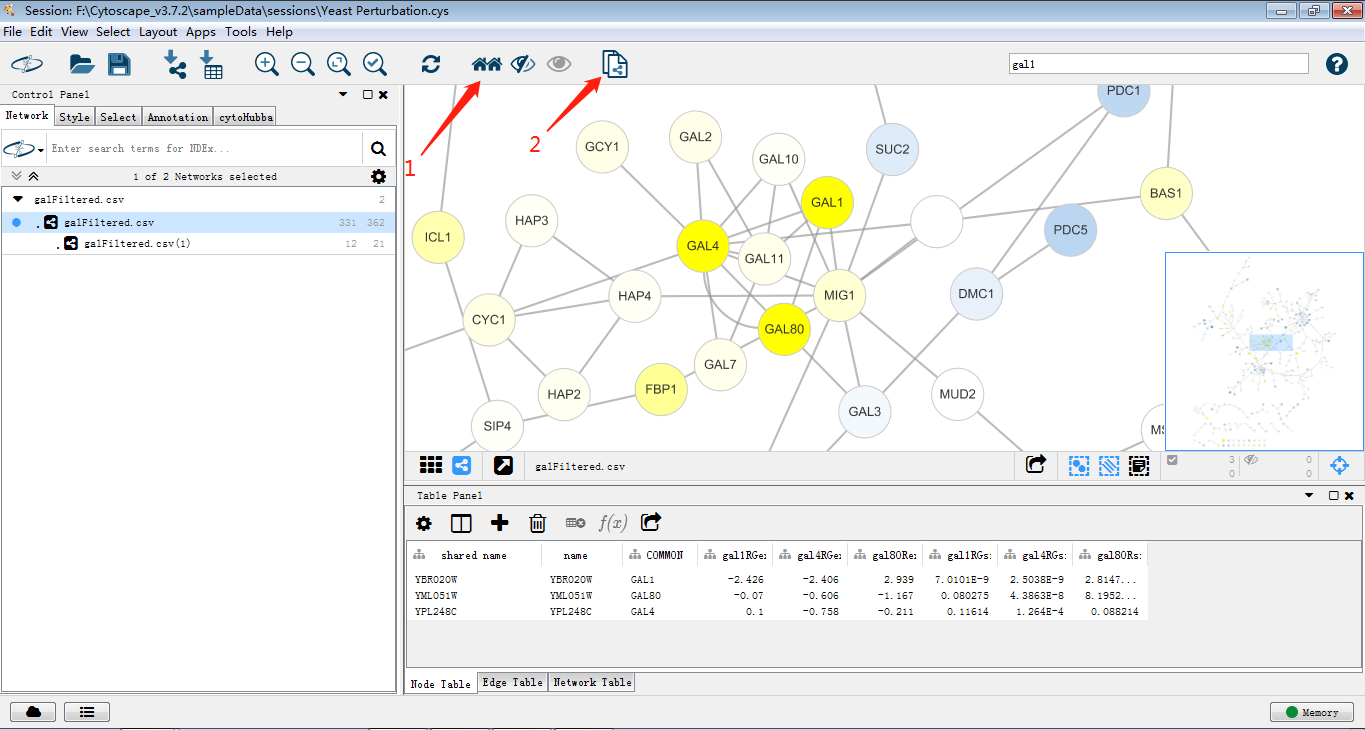
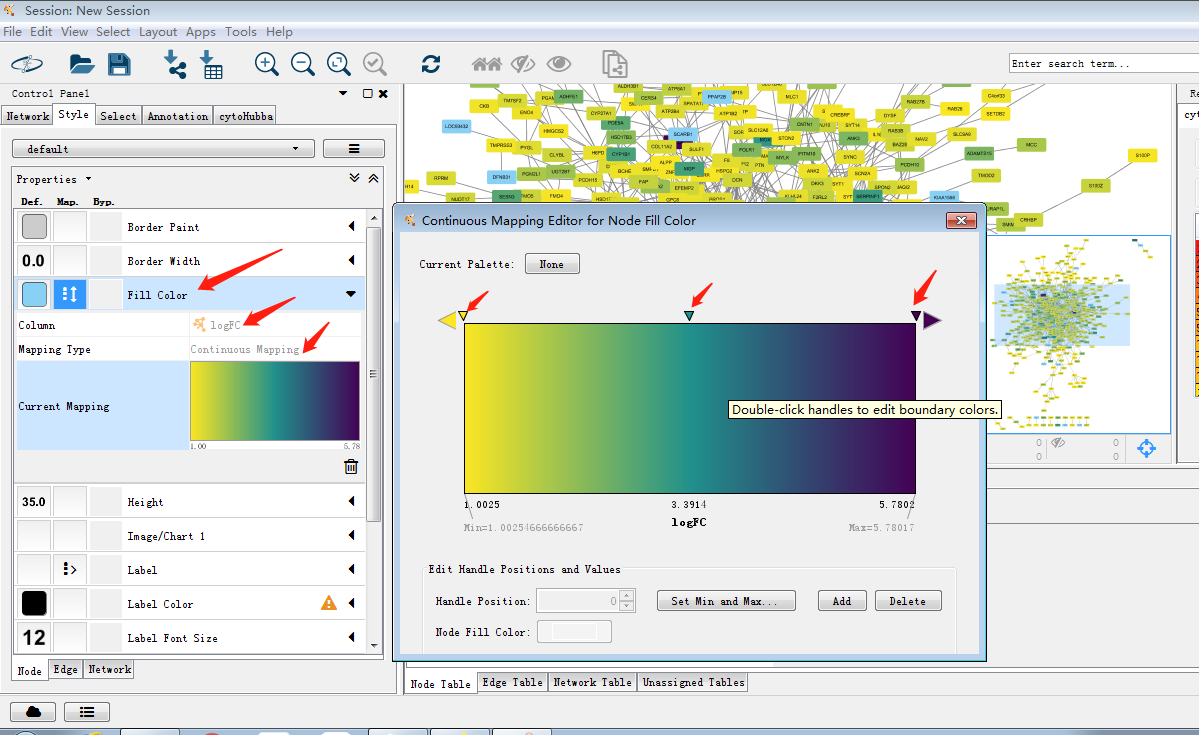
3.如果需要对图进行调整,可以使用控制面板中的style,这个后面详细说明。接下来使用cytohubba及MCODE插件对其进行模块筛选。这两个插件可以在界面左上角Apps中进行安装,JOJO在进行插件安装过程中碰到了网络问题,更新显示无法连接到APP store,这时可以选择去官网将插件下载下来再安装进cytoscape,但是JOJO秉着能偷懒绝不努力的原则手机开个热点电脑连接搞定╮(╯▽╰)╭
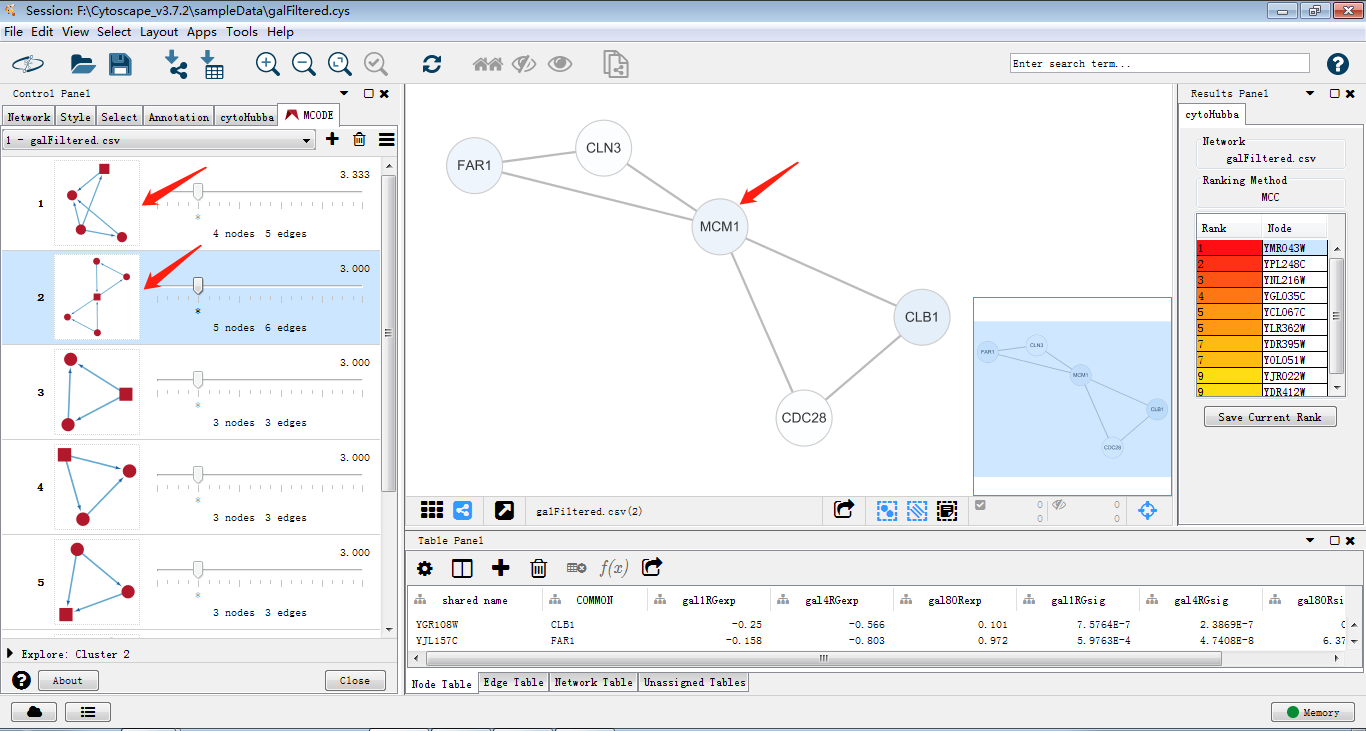
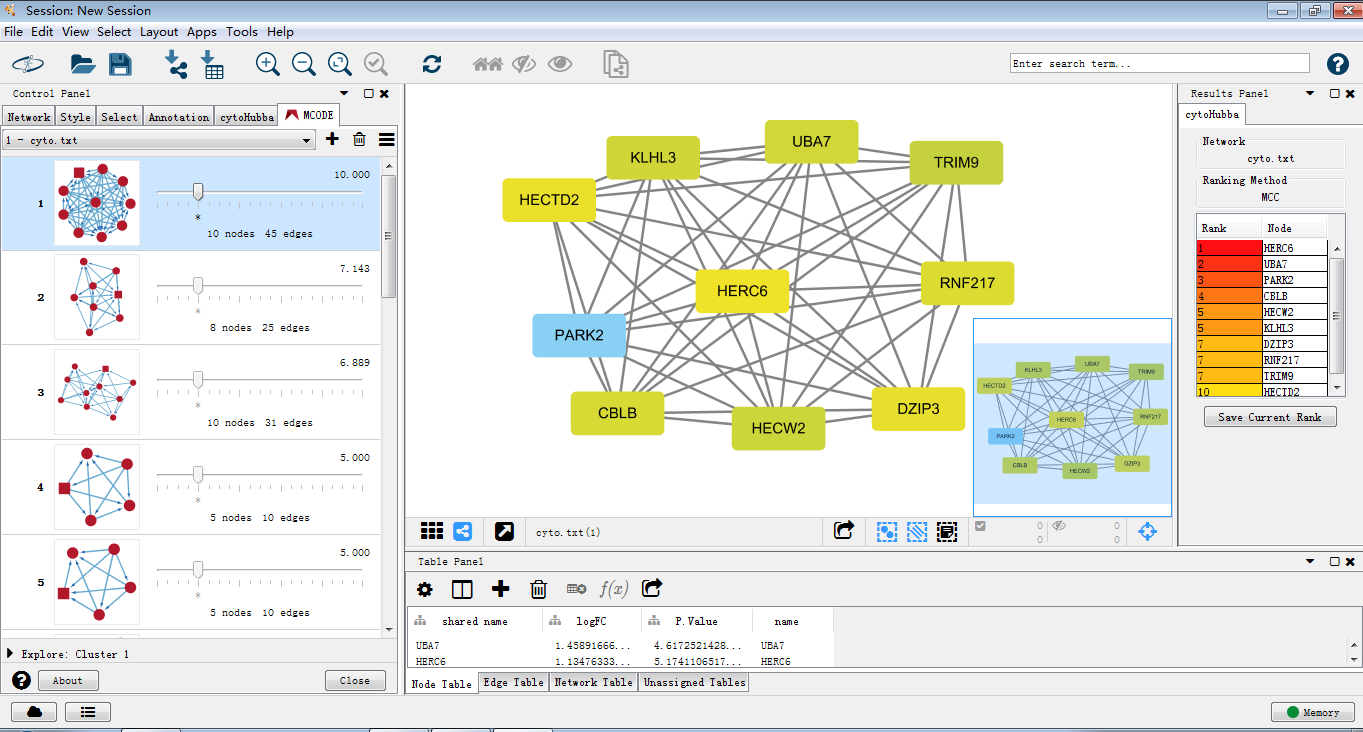
使用cytohubba把关键子网络提取出来,如图筛选出关键子网络,MCC是cytohubba的较新的算法,将lable换为common name,这样我们可以通过右侧的rank分看出MCM1在网络中占有重要权重以及其与GAL4的关系。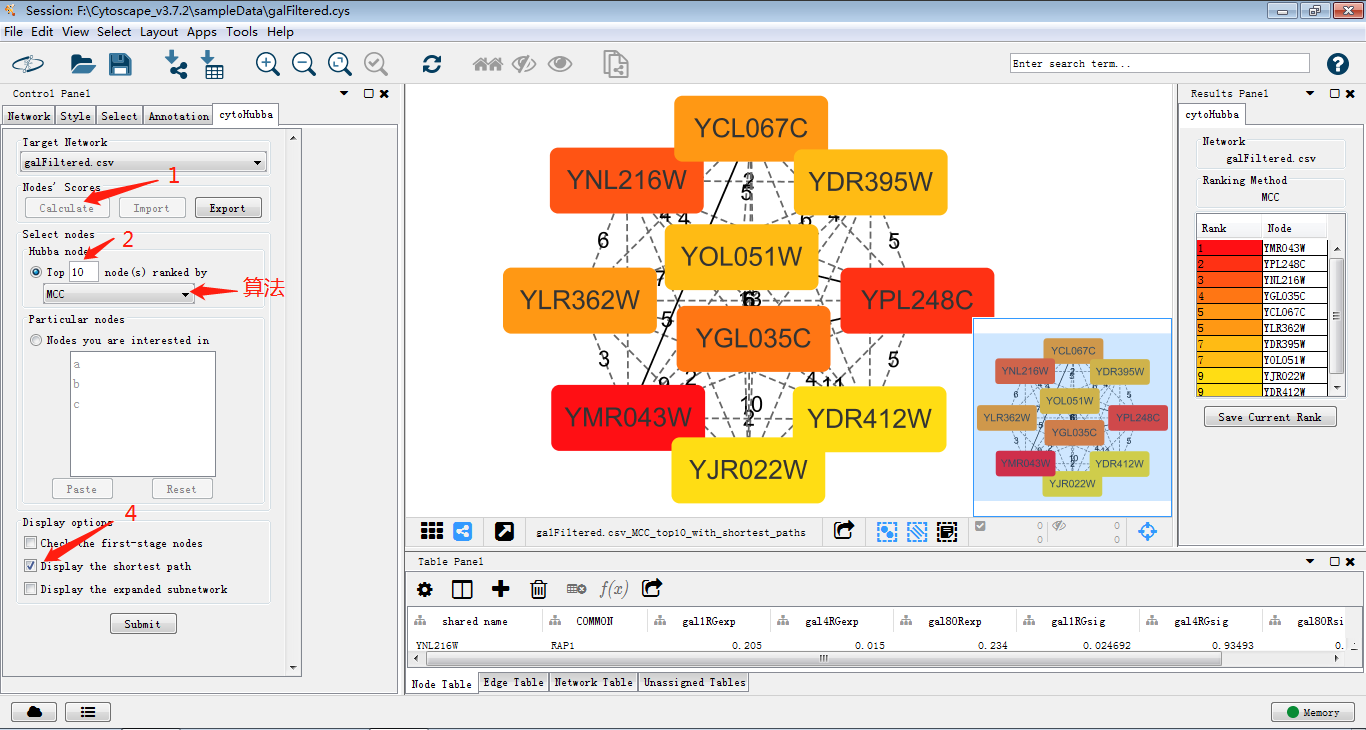
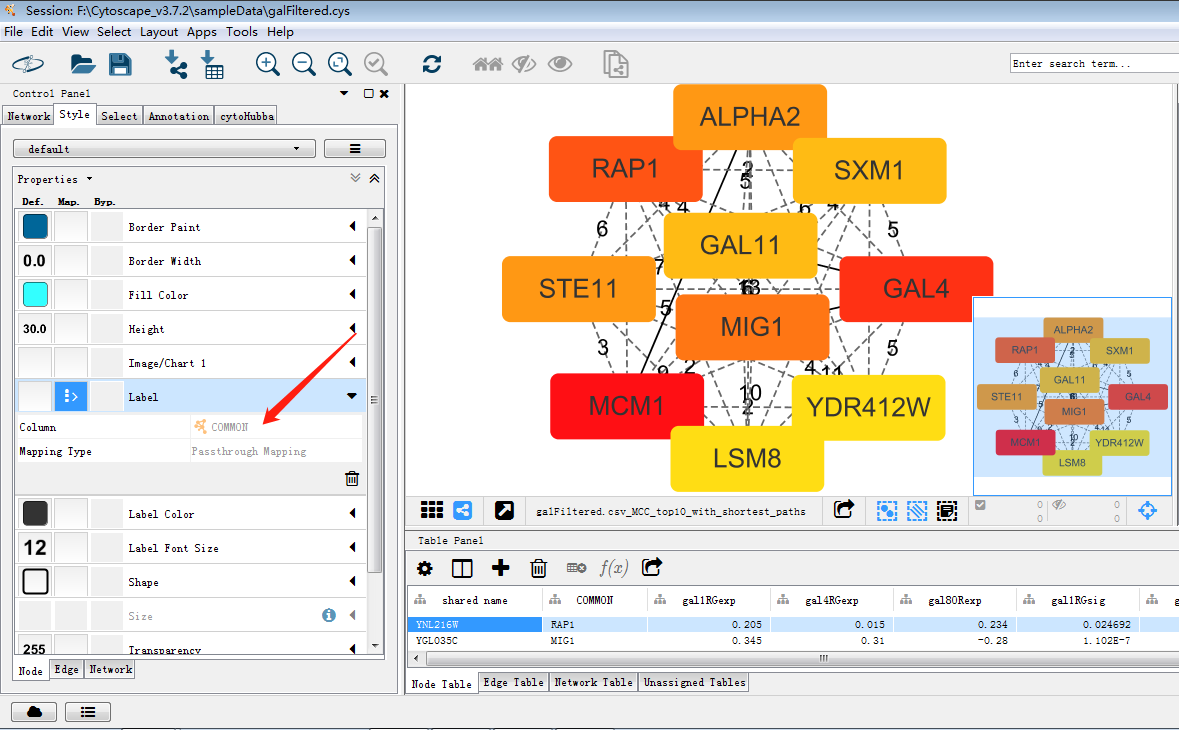
使用MCODE插件(以下引自https://www.jianshu.com/p/17a0ba0ced3c),
MCODE,Molecular COmplex Detection,发现PPI网络中紧密联系的regeions,这些区域可能代表分子复合体。根据给定的参数,分离dense regions,这相对其他cluster方法有其优点,因为其他的方法很少考虑网络的其余部分。总之MCODE可以发现PPI网络中相互作用的Dense region。这主要基于connection data,其中很多已经被证实是complex。这个函数不会被因高通量技术带来高假阳性影响。分子复合体预测很重要,因为这可以提供功能注释的另一个水平。因为sub-units of a molecular complex通常情况下,功能代表同一个生物目标分子,对一个未知pro的预测(作为复合体一部分),对这个pro的注释也增加了可信度。
JOJO觉得吧,这个MCODE就是一个算法工具,可以将网络图中的一些关键蛋白模块提取出来。直接使用默认参数提取模块,结果如下图,选取第1、2个模块出来反复鞭尸,可以发现第二个模块中的关键节点MCM1与上图中的关键节点是一致的,这意味着两种算法的得出的两种蛋白模块拥有相同的关键基因。那么这个基因与GAL1,GAL4,GAL80的表达关系就有待进一步探索了。实例分析:
通过以上,基本熟悉了cytoscape的操作,下面用GSE42872练习,使用R进行差异分析,获得差异基因,用差异基因制作string的输入数据,这个过程我们有标准代码,而且反复讲解了。走标准分析流程,火山图,热图,GO/KEGG数据库注释等等。这些流程的视频教程都在B站和GitHub了,目录如下:
- 第一讲:GEO,表达芯片与R
- 第二讲:从GEO下载数据得到表达量矩阵
- 第三讲:对表达量矩阵用GSEA软件做分析
- 第四讲:根据分组信息做差异分析
- 第五讲:对差异基因结果做GO/KEGG超几何分布检验富集分析
- 第六讲:指定基因分组boxplot指定基因list画热图
感兴趣可以细读表达芯片的公共数据库挖掘系列推文 ;- 解读GEO数据存放规律及下载,一文就够
- 解读SRA数据库规律一文就够
- 从GEO数据库下载得到表达矩阵 一文就够
- GSEA分析一文就够(单机版+R语言版)
- 根据分组信息做差异分析- 这个一文不够的
- 差异分析得到的结果注释一文就够
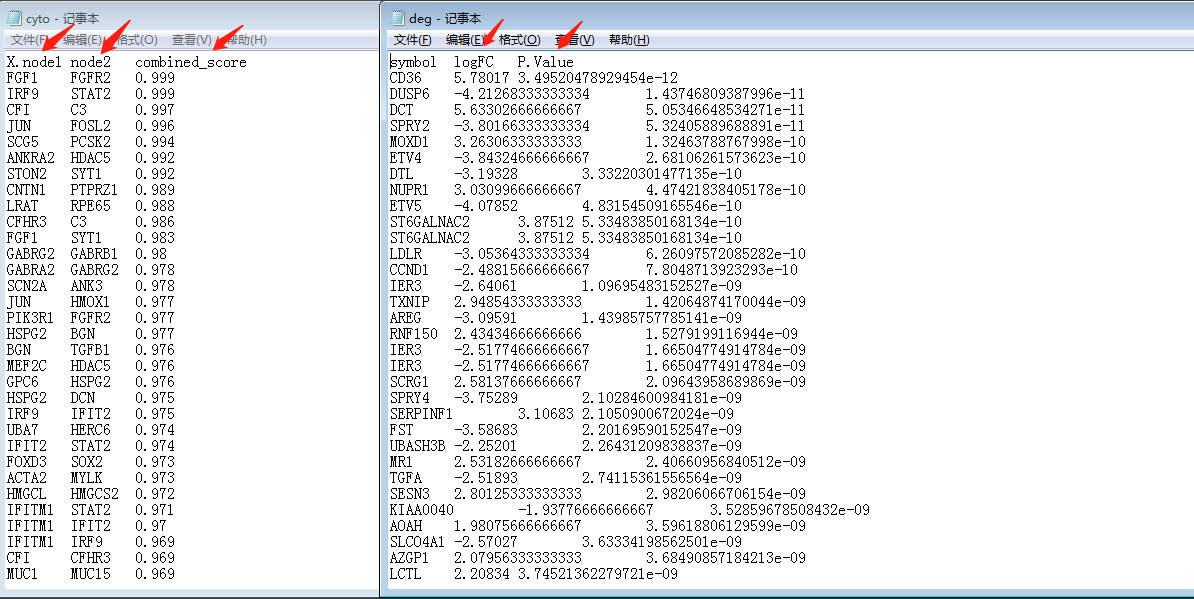
从string网页工具上下载string_interactions.tsv文件,再使用R对tsv文件进行初步处理成cytoscape的输入文件—cyto.txt 及 deg.txt,代码如下:#制作string的输入数据 load("step4output.Rdata") gene_up= deg[deg$change == 'up','symbol'] gene_down=deg[deg$change == 'down','symbol'] write.table(gene_up, file="upgene.txt", row.names = F, col.names = F, quote = F) write.table(gene_down, file="downgene.txt", row.names = F, col.names = F, quote = F) #准备cytoscape的输入文件 tsv = read.table("string_interactions.tsv",comment.char = "!",header = T) tsv2 = tsv[,c(1,2,ncol(tsv))] head(tsv2) write.table(tsv2, file = "cyto.txt", sep = "\t", quote = F, row.names = F) p = deg[deg$change != "stable",c("symbol","logFC","P.Value")] head(p) write.table(p, file = "deg.txt", sep = "\t", quote = F, row.names = F)老规矩,JOJO开始了!
1.先看数据,大致可以看出cyto.txt 中node1 node2即为节点, combined-score为节点互作关系,deg.txt为节点属性(基因表达量)。
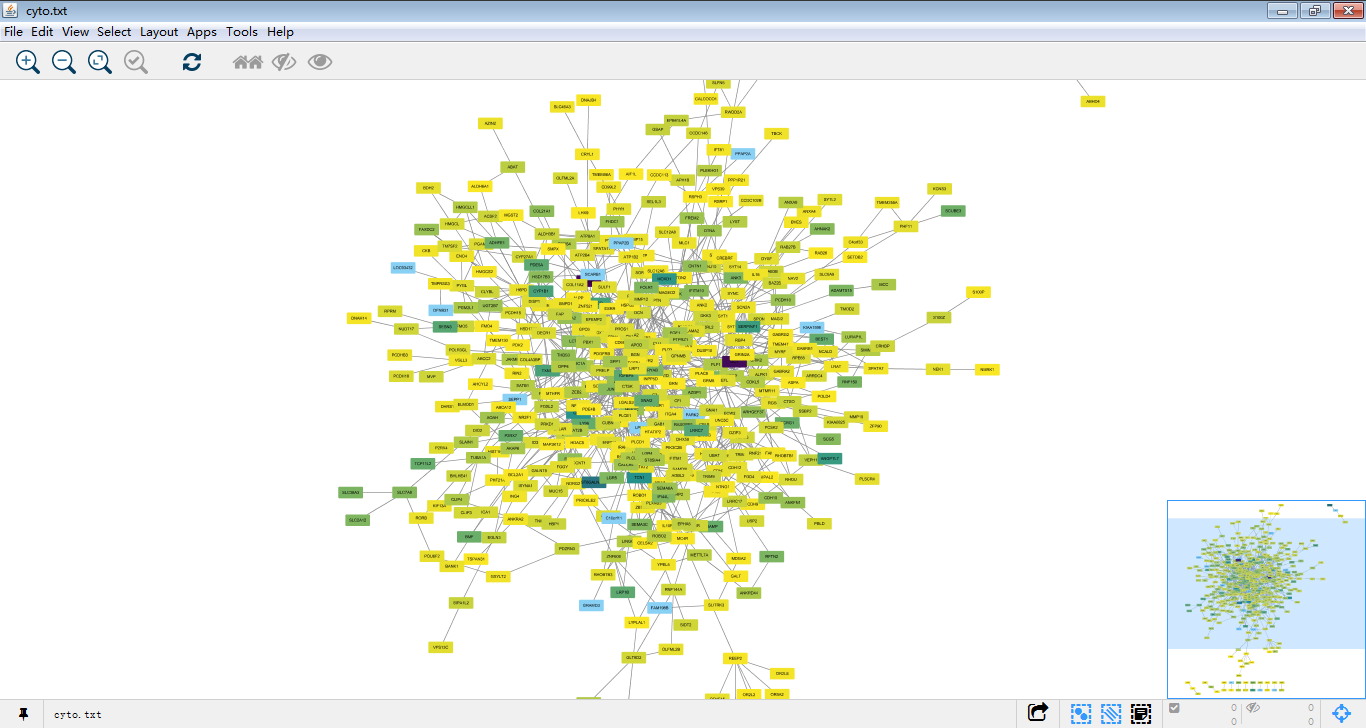
2.导入数据,
。。。诶呀,不对放错图了((・∀・)ゞ→→
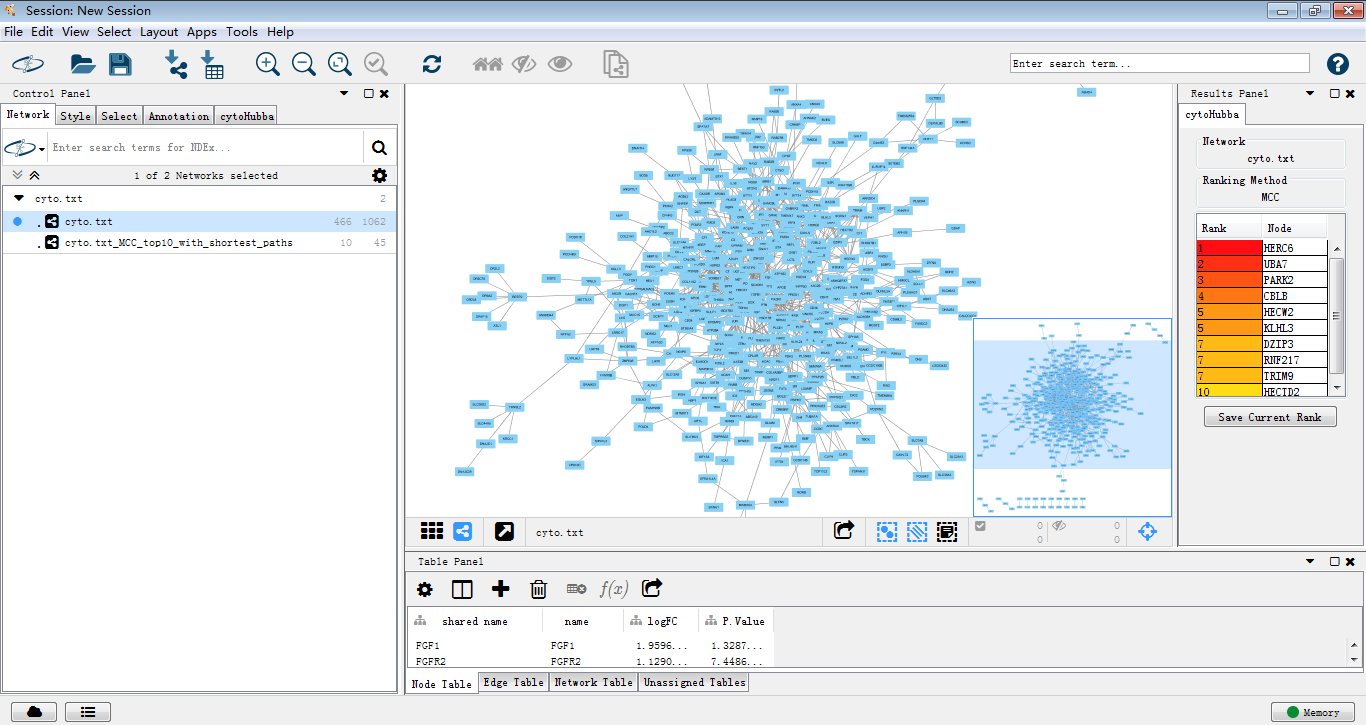
再来,
这才像话,,我们可以对这个图做一些美化(๑•̀ㅂ•́)و✧
同样,用MCODE处理后提取关键子网络。
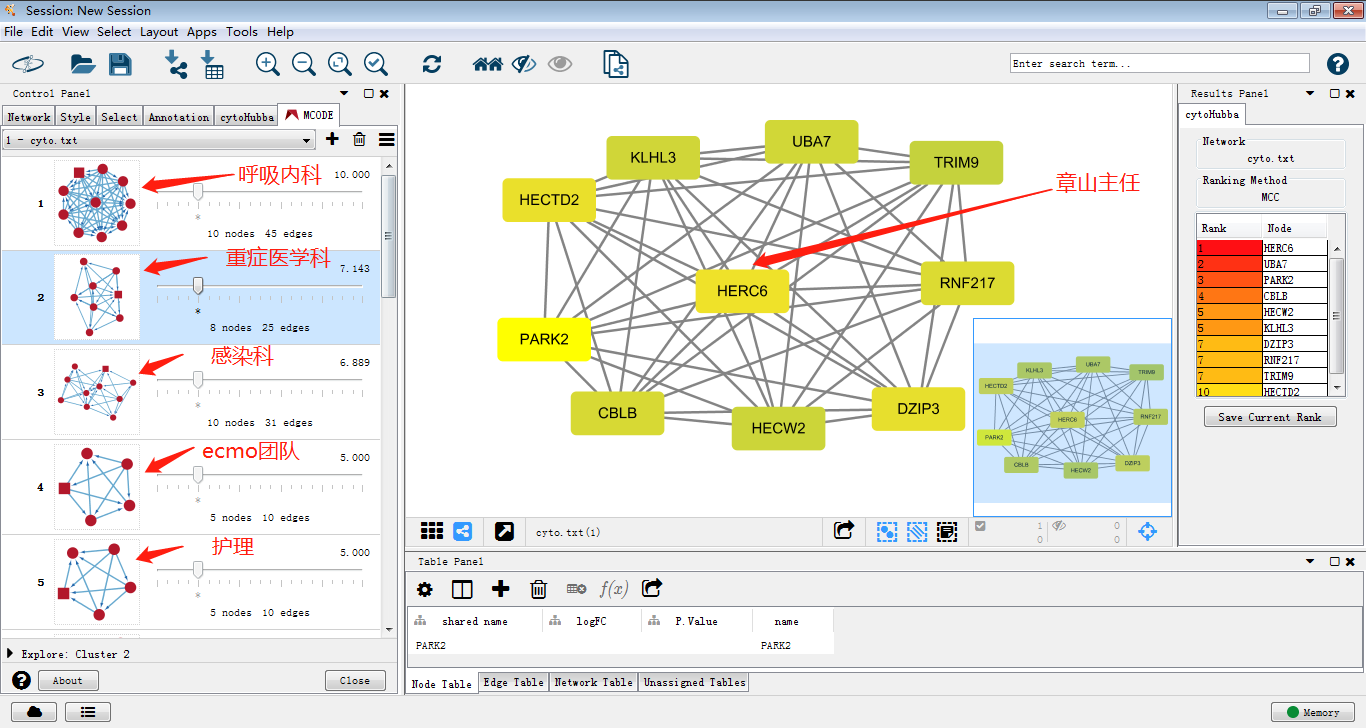
3.那么写了这么多,做了这么多,到底我们做出来的子网络是个什么呢,cytoscape给我们带来了什么便利?关于这点,*健明老师有一个形象的比喻便于理解。一起来理解子网络及hub基因
就好比这次新型冠状病毒疫情那么严重,其它省市兄弟医院从2万个医生中选出1000个医生支援武汉(差异分析提取差异基因1000个),但是如果新闻稿单单写1000个医生支援武汉未免显得单薄不能突出重点,于是就我们需要对这1000个医生进一步分析,他们都是来自哪些科室呢,由哪些团队构成呢(蛋白模块或者说子网络),于是我们调查(MCODE)发现这1000医生中有呼吸内科,重症医学科,感染科等等的科室构成,我们进一步了解,发现每个团队由一位经验最为丰富的领头人带队,呼吸内科带队的是章山主任,重症医学科带队的是李肆主任,他们领导的团队怎么样,他们在业界做了什么贡献,那么就可以写一篇完善的新闻稿了。
类比过来,我们从2万个基因中挑选出1000个差异基因,获得基因名(上调、下调),再从string把这些基因的相互作用关系拿出来,利用cytoscape做出网络图,用MCODE插件将其中的关键子网络挑选出来,选择感兴趣的基因做go分析,通路分析。
JOJO发现在读取数据的过程中偶有会有各种各样的问题,比如同样的读取方法同样的数据有时候会读取出空白圆形的图,有时候是蓝色方形。。。。**遇事不决,重启解决
参考:http://www.360doc.com/content/18/0410/10/42030643_744385648.shtml
http://www.360doc.com/content/17/0110/23/19913717_621651768.shtml
https://www.jianshu.com/p/17a0ba0ced3c