五年前我在生信菜鸟团博客写过一个《RNA-seq流程需要进化啦》,上面分享过:
Tophat 首次被发表已经是6年前
Cufflinks也是五年前的事情了
Star的比对速度是tophat的50倍,hisat更是star的1.2倍。
stringTie的组装速度是cufflinks的25倍,但是内存消耗却不到其一半。
Ballgown在差异分析方面比cuffdiff更高的特异性及准确性,且时间消耗不到cuffdiff的千分之一
Bowtie2+eXpress做质量控制优于tophat2+cufflinks和bowtie2+RSEM
Sailfish更是跳过了比对的步骤,直接进行kmer计数来做QC,特异性及准确性都还行,但是速度提高了25倍
kallisto同样不需要比对,速度比sailfish还要提高5倍!!!
如果你现在(2020)做人类数据分析,比如lncRNA的鉴定啥的,当然是走hisat2+stringTie流程啦,取代已经十多年了的tophat+Cufflinks流程。但是我这两天假期无聊刷文献,看到发表在Theranostics 2020,的研究文章:Long noncoding RNA PiHL regulates p53 protein stability through GRWD1/RPL11/MDM2 axis in colorectal cancer里面的RNA-seq数据居然还是在走十几年前的tophat流程哦,有趣,而且写的不清不楚那个FPKM是如何计算的。在广州锐博公司?

实际上,RNA-seq我们在生信技能树应该是至少推出了400篇教程,而且是我们全国巡讲的标准品知识点,其中还有一个阅读量过两万的综述翻译及其细节知识点的补充:
相信大家听完了我B站的RNA-seq分析流程后,对这个数据的应用方向都不陌生。代码也很简单,如果你有Linux基础,基本上一两个小时就可以完成数据分析流程,拿到表达矩阵啦。就是:
# 安装RNA-seq数据处理流程
# 代码参考:https://www.jianshu.com/p/a84cd44bac67
# 视频教程见:https://www.bilibili.com/video/av28453557
hisat2=/home/jianmingzeng/biosoft/HISAT/hisat2-2.0.4/hisat2
# # 如果使用conda安装的 hisat2,那么 hisat2 命令应该是在环境变量的。
## 索引文件需要自己下载
# https://ccb.jhu.edu/software/hisat2/manual.shtml
# wget ftp://ftp.ccb.jhu.edu/pub/infphilo/hisat2/data/mm10.tar.gz
index=/home/jianmingzeng/reference/index/hisat/mm10/genome
ls raw_fq/*gz | while read id; do
$hisat2 -p 10 -x $index -U $id -S ${id%%.*}.hisat.sam
done
ls *.sam|while read id ;do (samtools sort -O bam -@ 5 -o $(basename ${id} ".sam").bam ${id});done
rm *.sam
ls *.bam |xargs -i samtools index {}
## gtf文件推荐去gencode数据库下载
gtf=/home/jianmingzeng/reference/gtf/gencode/gencode.vM12.annotation.gtf
featureCounts=/home/jianmingzeng/biosoft/featureCounts/subread-1.5.3-Linux-x86_64/bin/featureCounts
# # # 如果使用conda安装的 subread,那么featureCounts 命令应该是在环境变量的。
$featureCounts -T 5 -p -t exon -g gene_id -a $gtf -o all.id.txt *.bam 1>counts.id.log 2>&1 &
这篇文章其实并没有怎么使用这个RNA-seq数据,可能是因为确实他们课题组并不懂测序数据,也没有生物信息学基础知识背景吧,就是委托公司简单测序而已。
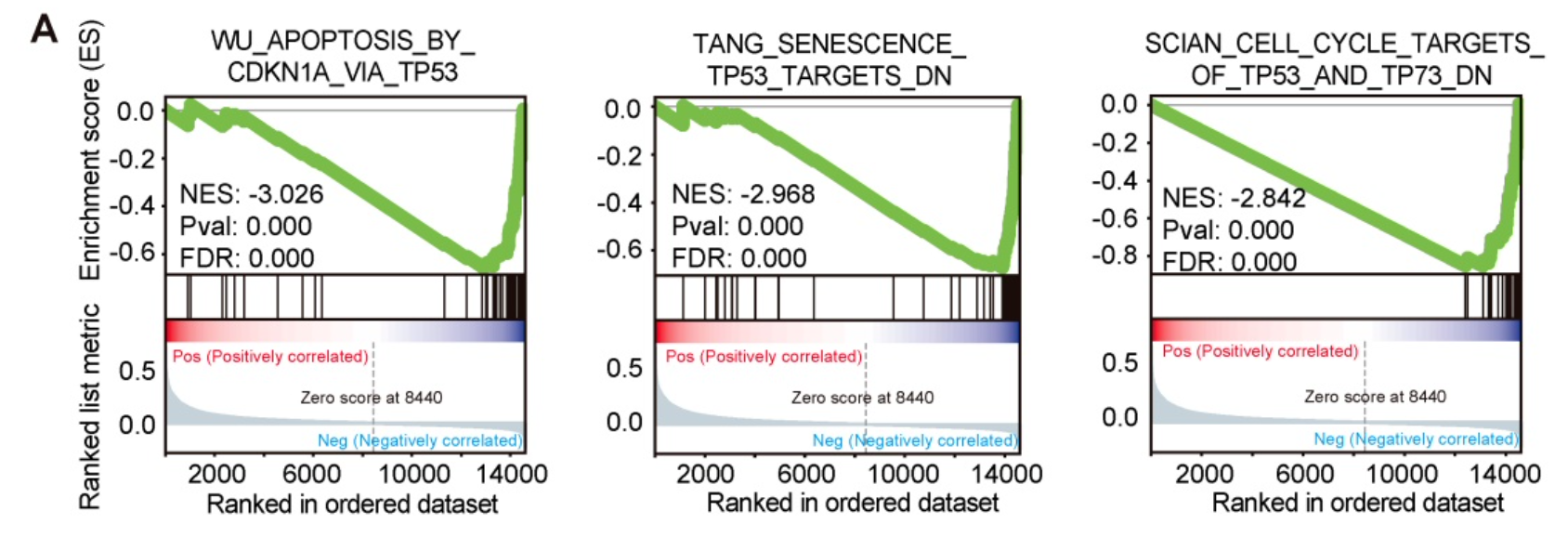
做的是GSEA分析
(A) Gene set enrichment analysis (GSEA) results based on PiHL expression levels (siRNA-PiHL vs siRNA-NC, with three repeats) in HCT116 cells.
The GSEA plots for the enrichment of p53 target genes involved in modulation of apoptosis and cell cycle are shown.
很简单的啦,
如果大家感兴趣GSEA分析原理和用法,看我在生信技能树的教程合辑
RNA-seq数据分析继续免费做
请注意,并不是上游流程哦,因为这个走hisat2+stringTie流程需要服务器,就有成本了。我们提供的免费分析,仅仅是我在生信技能树推文里面的各种各样的数据分析,那些都是我非常有经验的,比如我在lncRNA的一些基础知识 ,和lncRNA芯片的一般分析流程 介绍过的那些图表,以及下面的目录的分析内容 对我来说是举手之劳,希望可以帮助到你!
- 转录组数据分析的4个维度认识(数据分析继续免费哦) RNA-seq数据的2个分组差异分析,热图,PCA图,火山图等等
- 根据感兴趣基因看肝癌免疫微环境的T细胞亚群差异 条形图或者箱线图
- 查看感兴趣基因的甲基化水平和RNA表达水平(数据分析免费做)相关性 散点图或者箱线图
- 我不相信kmplot这个网页工具的结果(生存分析免费做)
- 单基因GSEA分析策略(数据分析免费做活动继续)
- 干扰一个基因然后分析全局基因表达其实是无法定位该基因完整功能(春节免费数据分析活动继续)
- log与否会改变rpkm形式表达矩阵top的mad基因列表 WGCNA分析免费做
- 甲基化信号值的差异分析也许不应该是看logFC 甲基化信号矩阵差异分析免费做
- WGCNA得到模块之后如何筛选模块里面的hub基因 WGCNA分析免费做
- 既然可以看感兴趣基因的生存情况,当然就可以批量做完全部基因的生存分析
还是老规矩,发送数据分析要求,以及简短的项目描述到我的邮箱 jmzeng1314@163.com 目前只接受邮件这个交流形式,谢谢合作,麻烦用心一点写!
邮件正文最好是加上你是啥时候认识生信技能树的哦,或者其它一些寒暄的话,自我介绍也行。主要是考虑到可能想免费分析数据的朋友很多,所以会根据你的来信,我主观判定一个优先级哦。目前我有20多个愿意长期在我的指导下进行数据探索的学徒,等我的团队扩大到200人,我们应该是可以做到数据分析全部免费,敬请期待哈!