最近在做illumina的甲基化芯片数据处理知识整理,所以看了不少文献咯,发现甲基化探针到底需要进行哪些过滤这么一个小细节,值得探讨的却很多。欢迎大家加入我们的讨论群:一个甲基化芯片信号值矩阵差异分析的标准代码 ,我整理的资料,都在群里共享。
比如文献 A Minimal DNA Methylation Signature in Oral Tongue Squamous Cell Carcinoma Links Altered Methylation with Tumor Attributes. Mol Cancer Res 2016 Sep 里面居然特意去把450K芯片的45万个探针的碱基序列,拿去比对,然后过滤了近2万个不能唯一比对到参考基因组的探针。

实际情况下,能下载450K芯片的45万个探针,并且做碱基序列的科研人员很少,但是大家又急需处理它,所以可以使用minfi或者champ的成熟的甲基化探针过滤流程,下面我们一一讲解。
minfi可以把450K过滤到420K
一般来说的过滤,就是minfi包流程,你看到的甲基化芯片数据处理文章都会简要的在methods部分写一下过程,如下:

代码如下,首先你需要参考教程 甲基化芯片数据的一些质控指标 ,把甲基化芯片项目的每个样本的idat原始文件读入R里面成为 rgSet 这个变量,这个变量是对象 class: RGChannelSet ,可以使用minfi包进行各种各样的过滤哈。
看起来代码很长,实际上就对应着上面文献描述的过滤流程:
- 首先去除低质量甲基化芯片的探针
- 然后去除性染色体上面的探针
- 再去除一些reactive probes 或者多态性snp位点。
detP <- detectionP(rgSet)
head(detP)
mSetSq <- preprocessQuantile(rgSet)
detP <- detP[match(featureNames(mSetSq),rownames(detP)),]
keep <- rowSums(detP < 0.01) == ncol(mSetSq)
table(keep)
# 可以看到,这个步骤过滤的探针很有限
mSetSqFlt <- mSetSq[keep,]
mSetSqFlt
# 继续过滤性别相关探针,非常重要
ann450k <- getAnnotation(IlluminaHumanMethylation450kanno.ilmn12.hg19)
head(ann450k)
keep <- !(featureNames(mSetSqFlt) %in% ann450k$Name[ann450k$chr %in%
c("chrX","chrY")])
table(keep)
mSetSqFlt <- mSetSqFlt[keep,]
# remove probes with SNPs at CpG site
mSetSqFlt <- dropLociWithSnps(mSetSqFlt)
mSetSqFlt
# BiocManager::install("methylationArrayAnalysis",ask = F,update = F)
dataDirectory <- system.file("extdata", package = "methylationArrayAnalysis")
# list the files
# exclude cross reactive probes
xReactiveProbes <- read.csv(file=paste(dataDirectory,
"48639-non-specific-probes-Illumina450k.csv",
sep="/"), stringsAsFactors=FALSE)
keep <- !(featureNames(mSetSqFlt) %in% xReactiveProbes$TargetID)
table(keep)
mSetSqFlt <- mSetSqFlt[keep,]
mSetSqFlt
# 全部过滤完成后,还剩下420K的甲基化位点
champ可以把450K过滤到400K
champ过滤流程就没有那么多代码了,在读入甲基化芯片项目的每个样本的idat原始文件或者项目信号值矩阵的时候就自动过滤。
默认就做了6个步骤的过滤,如下:
- First filter is for probes with detection p-value (default > 0.01).
- Second, ChAMP will filter out probes with <3 beads in at least 5% of samples per probe.
- Third, ChAMP will by default filter out all non-CpG probes contained in your dataset.
- Fourth, by default ChAMP will filter all SNP-related probes.
- Fifth, by default setting, ChAMP will filter all multi-hit probes.
- Sixth, ChAMP will filter out all probes located in chromosome X and Y.
代码如下,同样是需要参考教程 甲基化芯片数据的一些质控指标 ,保证当前工作目录下面有idat文件夹,里面有idat文件哈,还需要自己制作一个csv的样本信息表格,非常耗费时间精力的, 但是不得不做。
myLoad <- champ.load('idat/',arraytype="450K")
save(myLoad,file = 'GSE68777_champ_load.Rdata')
如果是甲基化信号值矩阵,直接走 champ.filter 流程即可,难度在于信号值矩阵文件导入R里面的操作:
rm(list = ls())
options(stringsAsFactors = F)
require(GEOquery)
require(Biobase)
library("impute")
info=read.table("group.txt",sep="\t",header=T)
library(data.table)
a=fread("data.txt",data.table = F )
a[1:4,1:4]
rownames(a)=a[,1]
a=a[,-1]
beta=as.betarix(a)
beta=impute.knn(beta)
betaData=beta$data
betaData=betaData+0.00001
a=betaData
a[1:4,1:4]
identical(colnames(a),rownames(b))
library(ChAMP)
# beta 信号值矩阵里面不能有NA值
myLoad=champ.filter(beta = a,pd = b)
save(myLoad,file = 'step1-output.Rdata')
为什么会有那么多过滤步骤呢
其实你可以一步步检查,过滤掉的那些探针是否影响你的甲基化芯片项目数据分析结果,如果你时间足够的话,比如我就探索了性染色体的。如下:
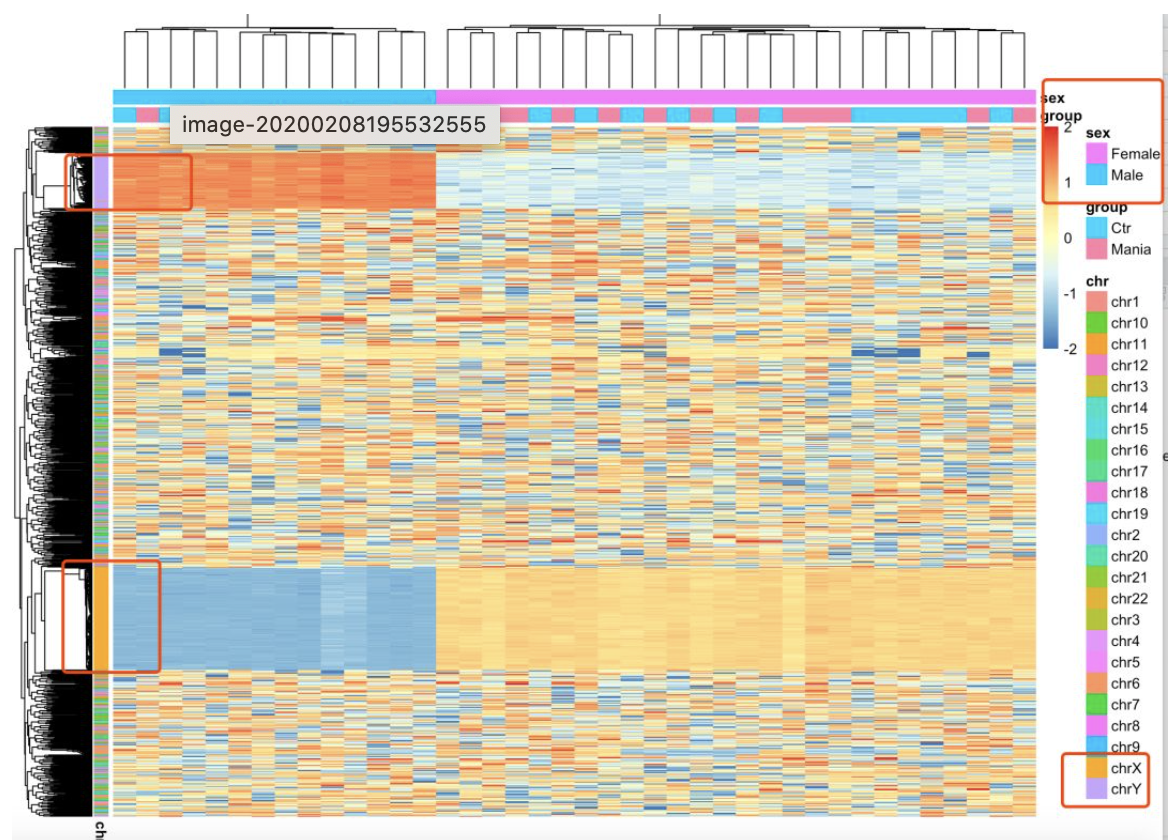
可以很明显的看到,top1000的sd的甲基化探针,主要是在性染色体上面差异巨大,如果这些性染色体的探针不去掉,你找差异甲基化探针就会被它干扰啦!
话说,为什么当初illumina在设计450K的时候就忽略这50K的探针呢?这不是浪费资源吗?不过,也许当初illumina公司的研发人员出发点是好的,可是后来研究人员在探讨450K数据处理的时候,慢慢形成的标准流程吧。
文末友情宣传
强烈建议你推荐给身边的博士后以及年轻生物学PI,多一点数据认知,让他们的科研上一个台阶:
-
全国巡讲全球听(买一得五) ,你的生物信息学入门课
-
生信技能树的2019年终总结 ,你的生物信息学成长宝藏
- 2020学习主旋律,B站74小时免费教学视频为你领路