现在一起看看拿到表达矩阵后有一个很重要的质控过程,就是根据一些阈值来过滤不合格细胞和基因。细胞的不合格很容易理解, 可能是那个细胞检测到的基因数量太少,或者太多,也许是那个细胞的文库大小异常,都是需要谨慎考虑是否管理它。
基因的不合格,就需要大家对人类参考基因组的注释信息有所背景了。走RNA-seq定量流程,拿到的表达矩阵通常是取决于gtf文件的注释程度,人类的gtf里面五万多个基因,不可能都在你的单细胞转录组项目数据里面出现。
首先细胞的取舍主要是看文库大小和检测到的基因数量
单细胞转录组通常使用10x数据,所以细胞数量惊人,每个样本可以是3000到10000的细胞数量都可以,取决于实验设计。每个细胞平均可以是1到10万的reads文库,都没有问题。
那么,每个细胞可以检测到多少基因数量呢,就取决于每个细胞分到的reads总数,1到10万的reads文库对应着200到1000的基因数量。
但是Smart-seq2技术的单细胞数据就不一样了,每个样本细胞数量通常是96的倍数,500个左右就很厉害了,然后每个细胞的reads就可以很多,百万级别都没有问题。所以检测到基因数量也很多,如下:

所以在你设置阈值来过滤不合格细胞的时候,一定要先理解好你的单细胞转录组数据,给全部的细胞检测到的基因数量绘制一个boxplot,看看哪些细胞所检测到的基因数量偏多或者偏少,一般来说,这样的的离群细胞就是你需要去除的!比如我在单细胞转录组教学就介绍过一个代码:
box <- lapply(colnames(sample_ann[,1:19]),function(i) {
dat <- sample_ann[,i,drop=F]
dat$sample=rownames(dat)
## 画boxplot
ggplot(dat, aes('all cells', get(i))) +
geom_boxplot() +
xlab(NULL)+ylab(i)
})
plot_grid(plotlist=box, ncol=5 )
这样的话细胞的上游指标可以批量检验,如下:
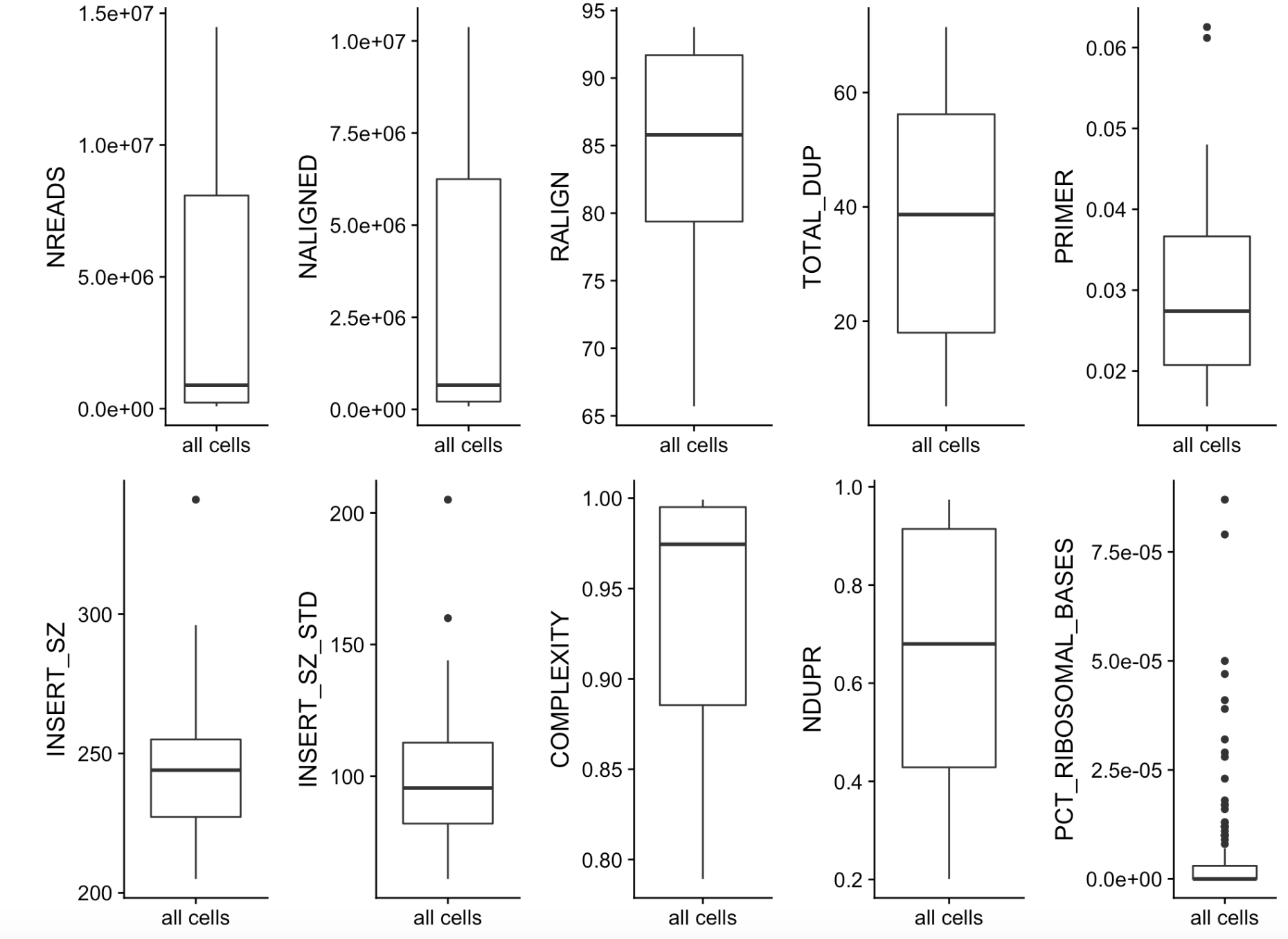
可以看到,每个细胞的测序文库大小和检测到基因数量虽然很重要,上游分析的其它质量指标,比对率,质量分数,duplicate情况,插入片段,内含子误差其实也会影响这个细胞的取舍。
然后是删除一些基因
前面我们提到过,gtf文件的注释程度不一样,拿到的表达矩阵通常是全部的基因,比如人类的gtf里面五万多个基因,你的表达矩阵就是5万多行。
实际上,大量的基因在你所有的细胞里面都是不表达的,这就是表达量全部为0的,肯定是需要去除啦。
还有一些基因仅仅是在5%左右的细胞表达,这个时候就很主观了,可能确实这个基因是那5%的稀有细胞的marker基因,也有可能就是单细胞转录组技术导致这个基因大量的drop-out了。
这个时候,你就需要卡一个阈值,到底一个基因表达多少算是表达呢,到底一个基因在多少个细胞里面有表达,你才保留下去,做下游分析呢?
后记
当然了,过滤细胞或者基因仅仅是单细胞转录组项目质量控制的开始,后面还有线粒体基因含量,核糖体基因含量,高表达量基因比例,细胞周期,批次效应,我们慢慢聊哈。
文末友情宣传
强烈建议你推荐给身边的博士后以及年轻生物学PI,多一点数据认知,让他们的科研上一个台阶:
- 全国巡讲全球听(买一得五) ,你的生物信息学入门课
- 生信技能树的2019年终总结 ,你的生物信息学成长宝藏
- 2020学习主旋律,B站74小时免费教学视频为你领路