前面我们一起学习了单细胞转录组数据的上游分析,而且了解了自己的项目的样本数量和测序量,还过滤了不合格的细胞和基因, 教程目录如下:
我们埋下了一个伏笔,就是拿到了差不多干净的表达矩阵后还有一个预处理步骤,但是这个步骤是选修,就是说你不会过滤线粒体核糖体基因也可以不做,没有人会责怪你,因为单细胞转录组数据分析本来就有难度。分析要点无穷无尽,即使你真的准备过滤线粒体核糖体基因你也会发现标准不好把握,需要看很多文献,还得结合你自己项目数据的实际情况啦。而且过滤线粒体核糖体基因并不是质控的终点,你还有细胞周期检查,单细胞活性检查,是否有两个细胞混在一起也是需要检查,非常的累。不仅仅是标准不好把握,而且因项目而异,具体情况具体判断。
处理线粒体核糖体基因的几个策略
粗暴的去除全部线粒体核糖体基因
直接在表达矩阵里面,去除掉属于的那一行表达量即可,有点类似于甲基化芯片数据分析,直接去除性染色体上面的全部探针,如下所示:

选择一个阈值来去除高表达量细胞
如下所示:

这个阈值很大程度上取决于你对自己项目的了解程度,不同器官组织提取的单细胞,本来就线粒体基因平均水平不一样, 不能一刀切!
这个只能是靠你看文献来获取认知了,多看多学习!
我的经验是,多次反复查看线粒体核糖体基因的影响,分群前后看,不同batch看,多次质控图表里面显示它,判断它是否是一个主要因素。
人类和小鼠的线粒体核糖体基因有哪些呢
我这里以gencode数据库的gtf文件为标准,在人类的gencode.v32.annotation.gtf 文件里面,可以查找到37个:
gene_id "ENSG00000210049.1"; gene_name "MT-TF"; hgnc_id "HGNC:7481"
gene_id "ENSG00000211459.2"; gene_name "MT-RNR1"; hgnc_id "HGNC:7470"
gene_id "ENSG00000210077.1"; gene_name "MT-TV"; hgnc_id "HGNC:7500"
gene_id "ENSG00000210082.2"; gene_name "MT-RNR2"; hgnc_id "HGNC:7471"
gene_id "ENSG00000209082.1"; gene_name "MT-TL1"; hgnc_id "HGNC:7490"
gene_id "ENSG00000198888.2"; gene_name "MT-ND1"; hgnc_id "HGNC:7455"
gene_id "ENSG00000210100.1"; gene_name "MT-TI"; hgnc_id "HGNC:7488"
gene_id "ENSG00000210107.1"; gene_name "MT-TQ"; hgnc_id "HGNC:7495"
gene_id "ENSG00000210112.1"; gene_name "MT-TM"; hgnc_id "HGNC:7492"
gene_id "ENSG00000198763.3"; gene_name "MT-ND2"; hgnc_id "HGNC:7456"
gene_id "ENSG00000210117.1"; gene_name "MT-TW"; hgnc_id "HGNC:7501"
gene_id "ENSG00000210127.1"; gene_name "MT-TA"; hgnc_id "HGNC:7475"
gene_id "ENSG00000210135.1"; gene_name "MT-TN"; hgnc_id "HGNC:7493"
gene_id "ENSG00000210140.1"; gene_name "MT-TC"; hgnc_id "HGNC:7477"
gene_id "ENSG00000210144.1"; gene_name "MT-TY"; hgnc_id "HGNC:7502"
gene_id "ENSG00000198804.2"; gene_name "MT-CO1"; hgnc_id "HGNC:7419"
gene_id "ENSG00000210151.2"; gene_name "MT-TS1"; hgnc_id "HGNC:7497"
gene_id "ENSG00000210154.1"; gene_name "MT-TD"; hgnc_id "HGNC:7478"
gene_id "ENSG00000198712.1"; gene_name "MT-CO2"; hgnc_id "HGNC:7421"
gene_id "ENSG00000210156.1"; gene_name "MT-TK"; hgnc_id "HGNC:7489"
gene_id "ENSG00000228253.1"; gene_name "MT-ATP8"; hgnc_id "HGNC:7415"
gene_id "ENSG00000198899.2"; gene_name "MT-ATP6"; hgnc_id "HGNC:7414"
gene_id "ENSG00000198938.2"; gene_name "MT-CO3"; hgnc_id "HGNC:7422"
gene_id "ENSG00000210164.1"; gene_name "MT-TG"; hgnc_id "HGNC:7486"
gene_id "ENSG00000198840.2"; gene_name "MT-ND3"; hgnc_id "HGNC:7458"
gene_id "ENSG00000210174.1"; gene_name "MT-TR"; hgnc_id "HGNC:7496"
gene_id "ENSG00000212907.2"; gene_name "MT-ND4L"; hgnc_id "HGNC:7460"
gene_id "ENSG00000198886.2"; gene_name "MT-ND4"; hgnc_id "HGNC:7459"
gene_id "ENSG00000210176.1"; gene_name "MT-TH"; hgnc_id "HGNC:7487"
gene_id "ENSG00000210184.1"; gene_name "MT-TS2"; hgnc_id "HGNC:7498"
gene_id "ENSG00000210191.1"; gene_name "MT-TL2"; hgnc_id "HGNC:7491"
gene_id "ENSG00000198786.2"; gene_name "MT-ND5"; hgnc_id "HGNC:7461"
gene_id "ENSG00000198695.2"; gene_name "MT-ND6"; hgnc_id "HGNC:7462"
gene_id "ENSG00000210194.1"; gene_name "MT-TE"; hgnc_id "HGNC:7479"
gene_id "ENSG00000198727.2"; gene_name "MT-CYB"; hgnc_id "HGNC:7427"
gene_id "ENSG00000210195.2"; gene_name "MT-TT"; hgnc_id "HGNC:7499"
gene_id "ENSG00000210196.2"; gene_name "MT-TP"; hgnc_id "HGNC:7494"
所以你拿到自己的表达矩阵后,其实简单的看看基因名字是否以 MT- 开头即可哦。
所以你会看到seurat包的函数:PercentageFeatureSet 可以用来计算线粒体基因含量。
sce <- CreateSeuratObject(Read10X('../scRNA/filtered_feature_bc_matrix/'), "sce")
head(sce@meta.data)
GetAssayData(sce,'counts')
sce[["percent.mt"]] <- PercentageFeatureSet(sce, pattern = "^MT-")
VlnPlot(sce, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
上面的方法是修改 sce[[“percent.mt”]] ,下面我们演示 AddMetaData 函数,同样是可以增加线粒体基因含量信息到我们的seurat对象。
mt.genes <- rownames(sce)[grep("^MT-",rownames(sce))]
C<-GetAssayData(object = sce, slot = "counts")
percent.mito <- Matrix::colSums(C[mt.genes,])/Matrix::colSums(C)*100
sce <- AddMetaData(sce, percent.mito, col.name = "percent.mito")
sce[["percent.mito"]]
也可以是添加核糖体基因含量,同样的你需要知道核糖体基因的名字规则:
rb.genes <- rownames(sce)[grep("^RP[SL]",rownames(sce))]
percent.ribo <- Matrix::colSums(C[rb.genes,])/Matrix::colSums(C)*100
sce <- AddMetaData(sce, percent.ribo, col.name = "percent.ribo")
在人类的gencode.v32.annotation.gtf 文件里面,可以看到核糖体基因数量也不少哦。
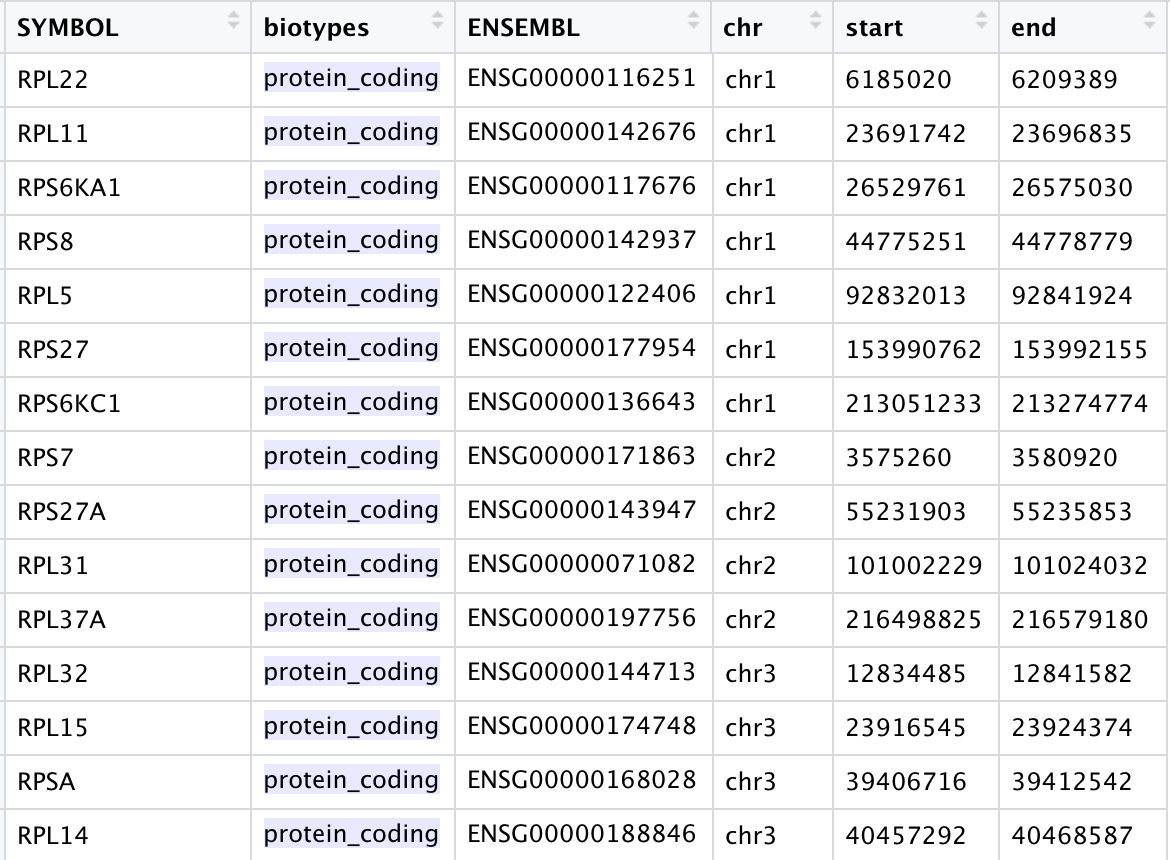
如果是小鼠,通常是基因名字大小写替换一下:
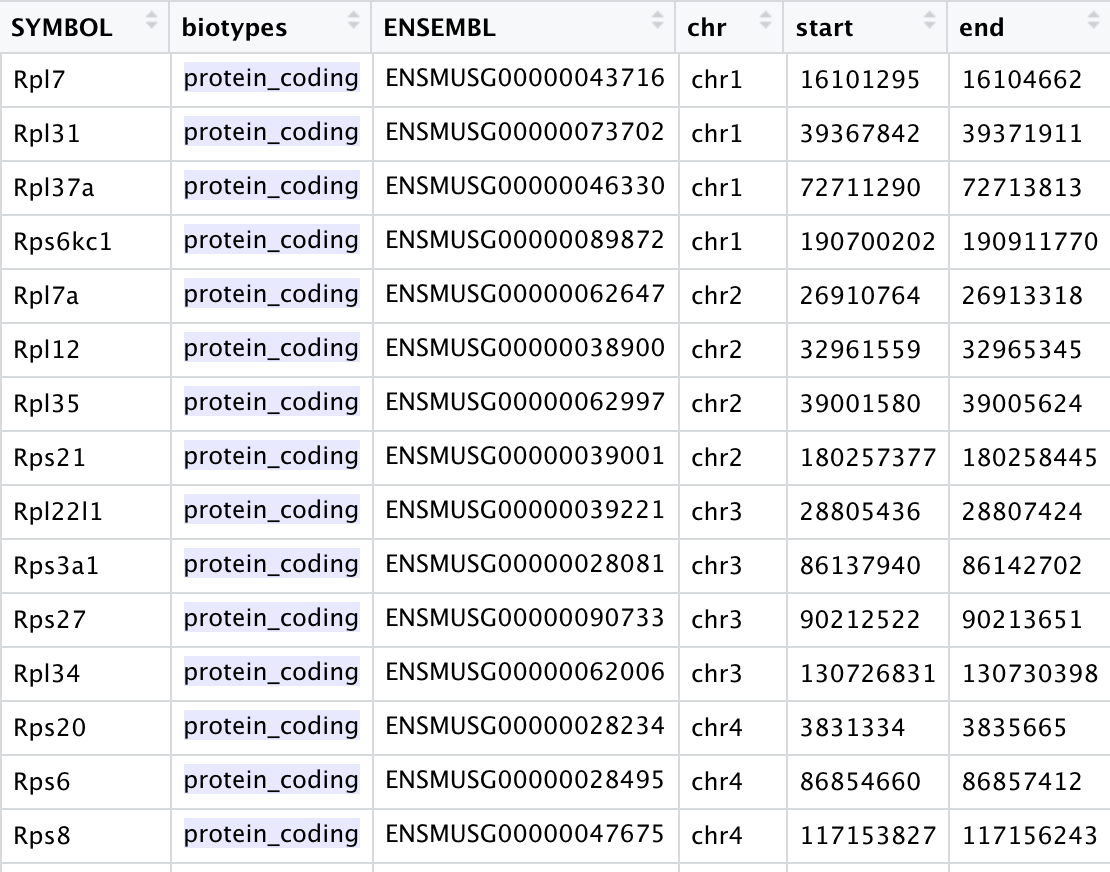
不仅仅是线粒体核糖体基因
还可以去免疫相关基因,缺氧相关基因,就更加的需要深入到你自己的课题,其实细节是无穷无尽的,但是我们的教学没办法做到如此的个性化,只能是精炼了常规单细胞转录组数据分析主线,就是5大R包, scater,monocle,Seurat,scran,M3Drop,然后10个步骤:
- step1: 创建对象
- step2: 质量控制
- step3: 表达量的标准化和归一化
- step4: 去除干扰因素(多个样本整合)
- step5: 判断重要的基因
- step6: 多种降维算法
- step7: 可视化降维结果
- step8: 多种聚类算法
- step9: 聚类后找每个细胞亚群的标志基因进行亚群命名
- step10: 继续分类
我们后面继续讲解质量控制的另外一个不容忽视的点,敬请期待。
文末友情宣传
强烈建议你推荐给身边的博士后以及年轻生物学PI,多一点数据认知,让他们的科研上一个台阶:
- 全国巡讲全球听(买一得五) ,你的生物信息学入门课
- 生信技能树的2019年终总结 ,你的生物信息学成长宝藏
- 2020学习主旋律,B站74小时免费教学视频为你领路