单细胞水平的研究是仅次于NGS的一次生物信息学领域的革命,同样的随随便便发CNS的黄金时期也过去了,现在想发高分文章,拿多个病人的多个样本进行单细胞转录组测序是非常正常的,比如下面的:
- 发表在 Nat Med. 2018 Aug,题目是:Phenotype molding of stromal cells in the lung tumor microenvironment, 共选取5例病人的共19个样本,通过10×genomics单细胞转录组测序探索基质细胞的亚群分类、基因功能(信号通路)、关键marker基因和临床预后,共鉴定出52个基质细胞亚群,
- 发表在 Nature Medicine (2018) ,标题是:Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis ,作者从3个乳腺癌患者体内通过FACS筛选到乳腺癌中肿瘤浸润淋巴细胞(TILs),使用商业仪器10X来做单细胞转录组,得到6,311个T细胞数据。
- 还有我们在单细胞天地分享的scRNA-seq揭示胰腺导管腺癌的瘤内异质性和恶性进展 ,有24个原发性PDAC肿瘤病人样本及11个对照胰腺样本(3例非胰腺肿瘤患者和8例非恶性胰腺肿瘤患者样本)
大多数百万经费起的项目,当然,现在想发普通的单细胞文章,也是得做多个样本了,就面临如何整合的问题,其中最出名的当然是Seurat包的CCA方法了,具体多火呢,发了才一年,引用就快破千!多个样本单细胞转录组数据整合算法
Seurat主要是处理10x单细胞转录组数据,而10x仪器商业上的成功可以说是成就了Seurat包,另外一个比较火的多个样本单细胞转录组数据整合算法是mutual nearest neighbors (MNNs)

当然,其它工具也有很多,我想你应该是不会看的,我就列出来而已: - MNNcorrect (https://doi.org/10.1038/nbt.4091)
- CCA + anchors (Seurat v3) (https://doi.org/10.1101/460147)
- CCA + dynamic time warping (Seurat v2) (https://doi.org/10.1038/nbt.4096) 今天介绍这个
- LIGER (https://doi.org/10.1101/459891)
- Harmony (https://doi.org/10.1101/461954)
- Conos(https://doi.org/10.1101/460246)
- Scanorama(https://doi.org/10.1101/371179)
- scMerge(https://doi.org/10.1073/pnas.1820006116)
Seurat关于多个单细胞转录组样本整合的文章实在是很厉害了,第一个发在Nature Biotechnology volume36, pages411–420 (2018),第二个发在CELL,Volume 177, Issue 7, 13 June 2019, 至少是我很长一段时间都无法企及的!
有趣的是 sctransform 还在预印本:Hafemeister, C. & Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. bioRxiv 576827 (2019). doi:10.1101/576827 不知道这个文章最后会在CNS的哪个子刊,或者干脆不发表了?
在CELL,Volume 177, Issue 7, 13 June 2019,文章里面描述了单细胞数据整合的两大问题:
- how can disparate single-cell datasets, produced across individuals, technologies, and modalities be harmonized into a single reference.
- once a reference has been constructed, how can its data and meta-data improve the analysis of new experiments?
示例的2个样本整合的效果
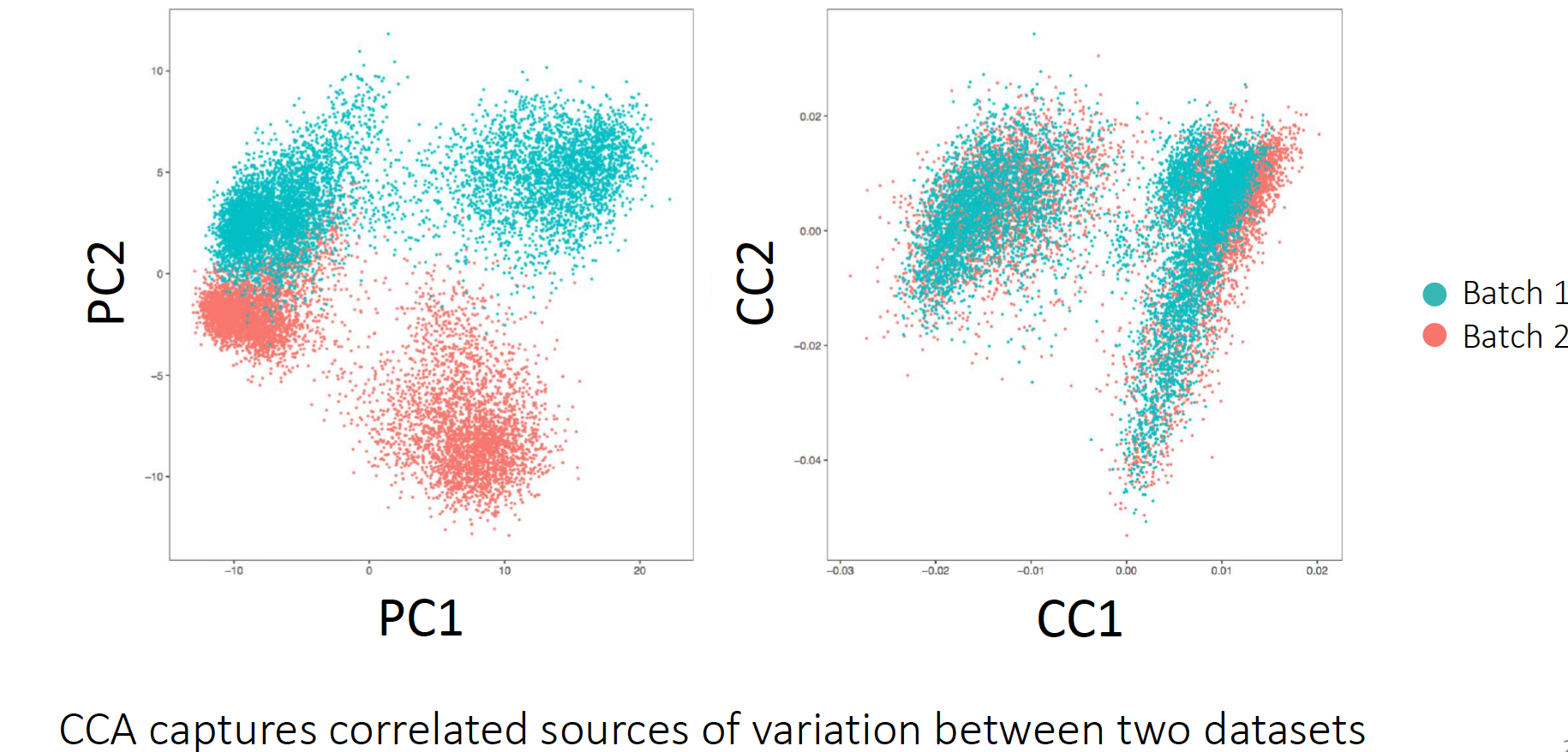
canonical correlation analysis (CCA)
在Seurat官网的最简单例子是两个样本,本来是有很明显的样本差异的,使用CCA整合后右图可以看到样本间差异就被抹去了。
如果你下载文章仔细学习,会发现作者还举了很多其它例子,包括不同单细胞转录组技术平台数据整合,甚至不同物种(人和鼠)的数据整合,还有不同物种不同技术平台的综合整合,可以说是很厉害了,如下:
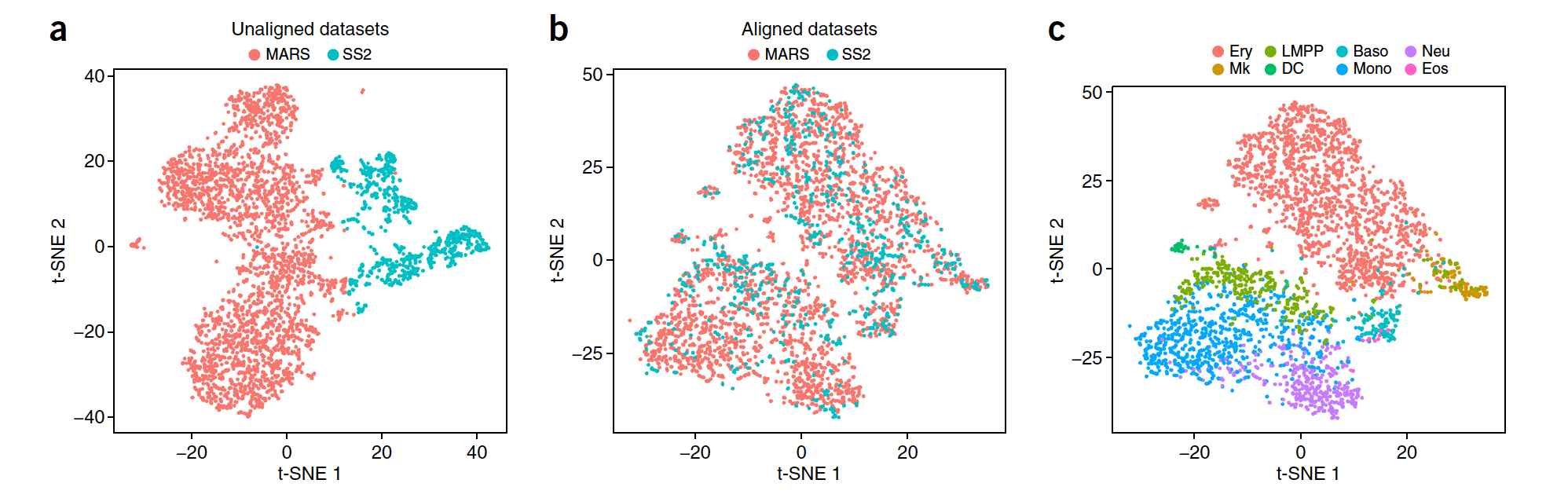
就是不同技术平台:3,451 hematopoietic progenitor cells from murine bone marrow sequenced using MARS-Seq (2,686) and SMART-Seq2 (SS2; 765).
如下:
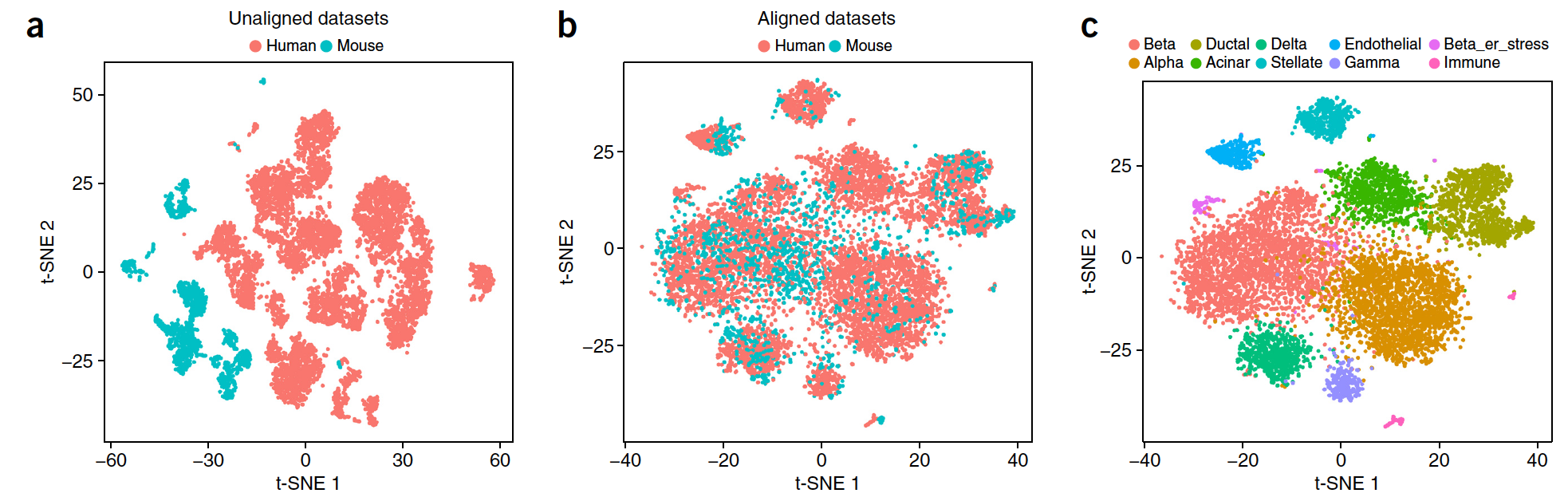
就是不同物种:10,191 pancreatic islet cells from human (n = 8,424 cells) and mouse (n = 1,767 cells) donors .用法代码
因为这个被他们实验室自己的CCA + anchors (Seurat v3)取代了,所以学这个CCA + dynamic time warping (Seurat v2) 的意义可能不大,我这里就贴一下作者的示例代码,来自于:https://rdrr.io/cran/Seurat/man/RunCCA.html 需要 (Seurat v3)
pbmc_small # As CCA requires two datasets, we will split our test object into two just for this example pbmc1 <- subset(pbmc_small, cells = colnames(pbmc_small)[1:40]) pbmc2 <- subset(pbmc_small, cells = colnames(x = pbmc_small)[41:80]) pbmc1[["group"]] <- "group1" pbmc2[["group"]] <- "group2" pbmc_cca <- RunCCA(object1 = pbmc1, object2 = pbmc2) # Print results print(x = pbmc_cca[["cca"]])不过,在单细胞天地我也会继续更新一下实际例子,测试数据在里面!

如果你需要这些单细胞转录组数据整合学习资料,可以去单细胞天地公众号回复数据整合,即可获取,其中10x数据上游处理都在:
- 单细胞实战(一)数据下载
- 单细胞实战(二) cell ranger使用前注意事项
- 单细胞实战(三) Cell Ranger使用初探
- 单细胞实战(四) Cell Ranger流程概览
- 单细胞实战(五) 理解cellranger count的结果
当然了,CCA + anchors (Seurat v3)的介绍更值得期待,同样是号称可以整合不同的 individuals, experimental conditions, technologies, or even species.