前面我们已经介绍了:使用monocle做拟时序分析(单细胞谱系发育) 然后回答了一个学员的问题: 拟时序分析的热图提取基因问题 ,但是因为大家对monocle包的说明书不熟悉,对R不熟练,以至于无法个性化处理monocle的各种中间结果,所以问题是多种多样。
同样的单细胞天地的基础视频课程学员提问:想知道参考文献的下面的条形图如何绘制,因为没有给原文,不知道作者定义的pseudotime bins是什么,不过在monocle官网教程,有一个state的概念,所以可以大致绘制出下面的图形:
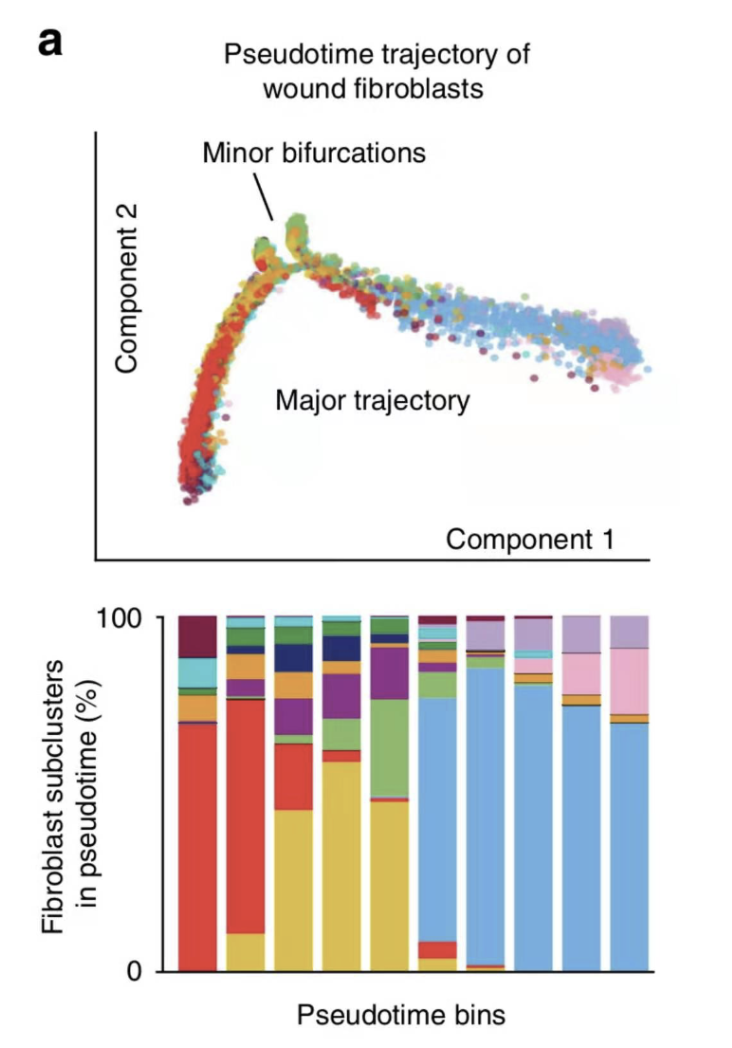
首先我们需要再次详细理解monocle输出的细胞谱系推断图
回到我们产生细胞谱系推断结果的函数:reduceDimension
load(file = 'ordering_genes_by_Biological_Condition_high.Rdata')
cds <- setOrderingFilter(cds, ordering_genes)
plot_ordering_genes(cds)
# 然后降维
cds <- reduceDimension(cds, max_components = 2,
method = 'DDRTree')
# 降维是为了更好的展示数据。
# 降维有很多种方法, 不同方法的最后展示的图都不太一样, 其中“DDRTree”是Monocle2使用的默认方法
# 接着对细胞进行排序
cds <- orderCells(cds)
## 最后两个可视化函数
plot_cell_trajectory(cds, color_by = "Biological_Condition")
实际上,就是给定有生物学意义的基因给 reduceDimension 函数,调整参数就可以出图,如下:
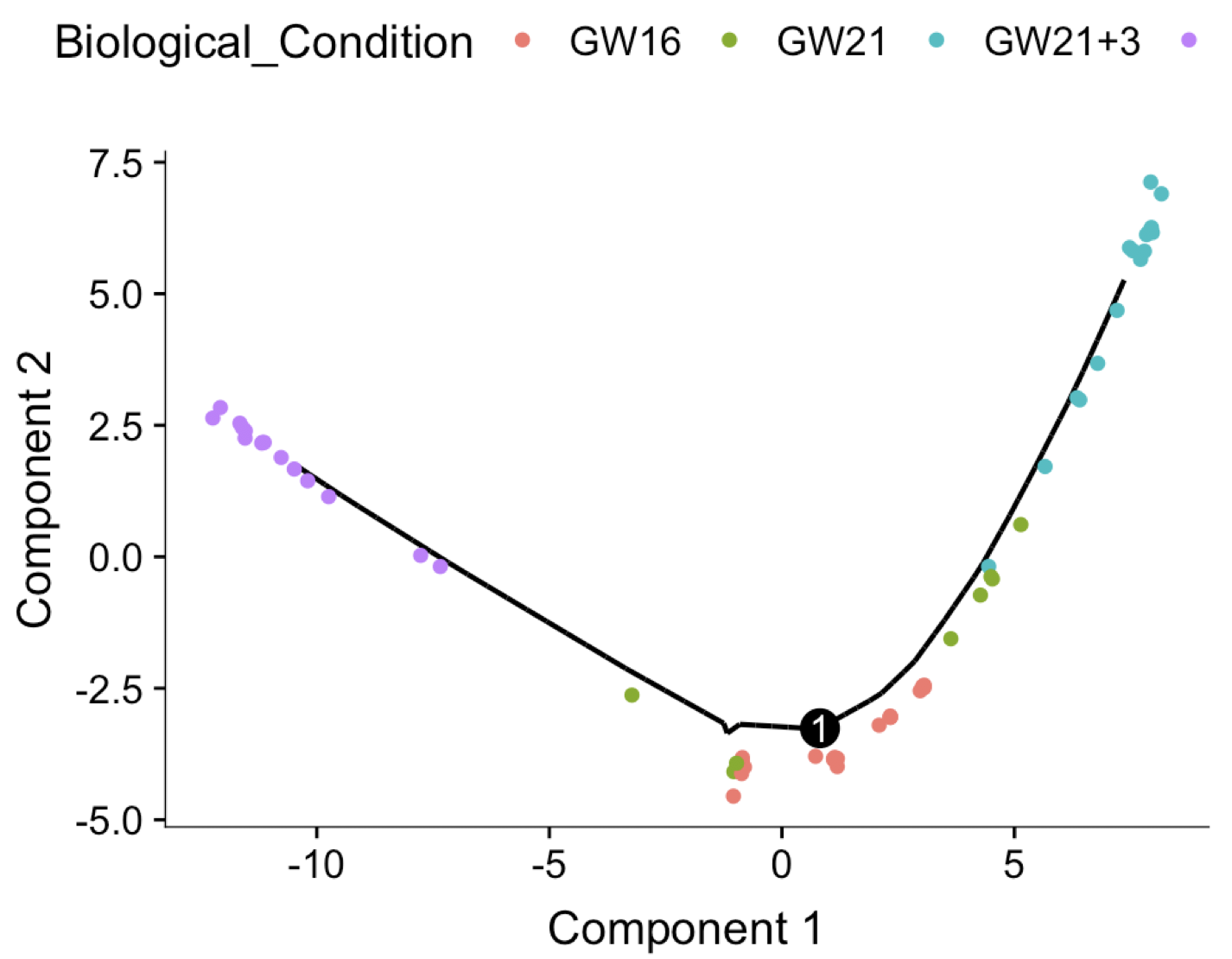
很容易理解,在这个细胞谱系推断图里面,其中一条轨迹是真实存在的,就是“GW16” and “GW21” ,“GW21+3” 这种孕期细胞,本身就是具有时间先后属性的,而且我们的monocle也的确找到了,在这个测试数据。
仔细摸索 reduceDimension 函数返回值
当然是先看函数的帮助文档了,在构建细胞谱系的时候有两个算法可以选择,而且还可以加入已知的混杂因素,这样reduceDimension 函数会帮助进行去混杂因素,做纯粹的细胞谱系推断
Monocle provides two different algorithms for dimensionality reduction via reduceDimension. Both take a CellDataSet object and a number of dimensions allowed for the reduced space. You can also provide a model formula indicating some variables (e.g. batch ID or other technical factors) to "subtract" from the data so it doesn't contribute to the trajectory.
但是,帮助文档里面更多是对参数的介绍,帮助我们运行reduceDimension 函数,并没有对它返回值的介绍,需要我们自行摸索了。我们 str(cds) 就可以看到的确是这个对象被这个函数修改了,添加了两列信息,如下:

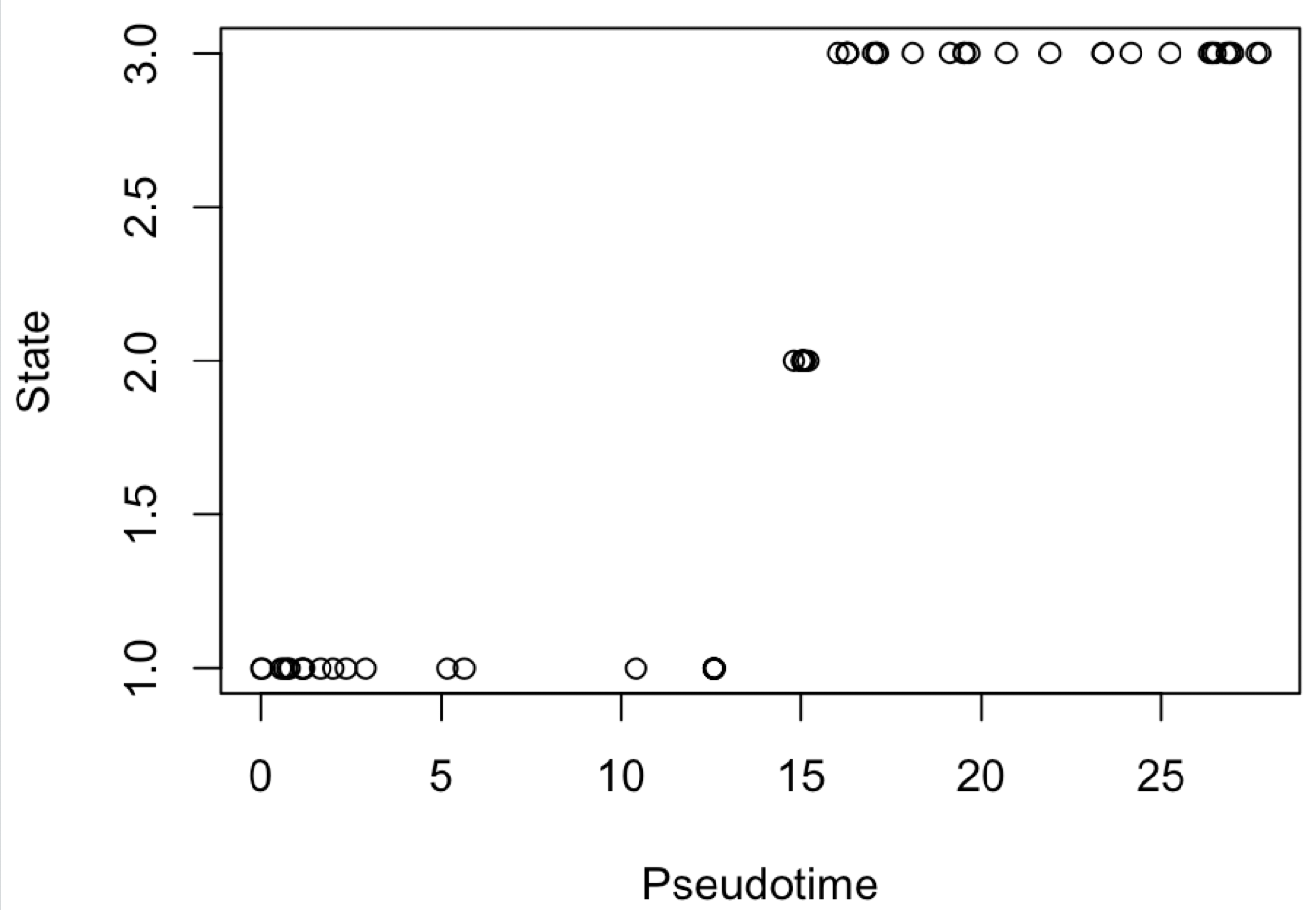
我们同样是简单探索一下Pseudotime和State的关系:
tmp=phenoData(cds)@data
plot(tmp[,30:31])
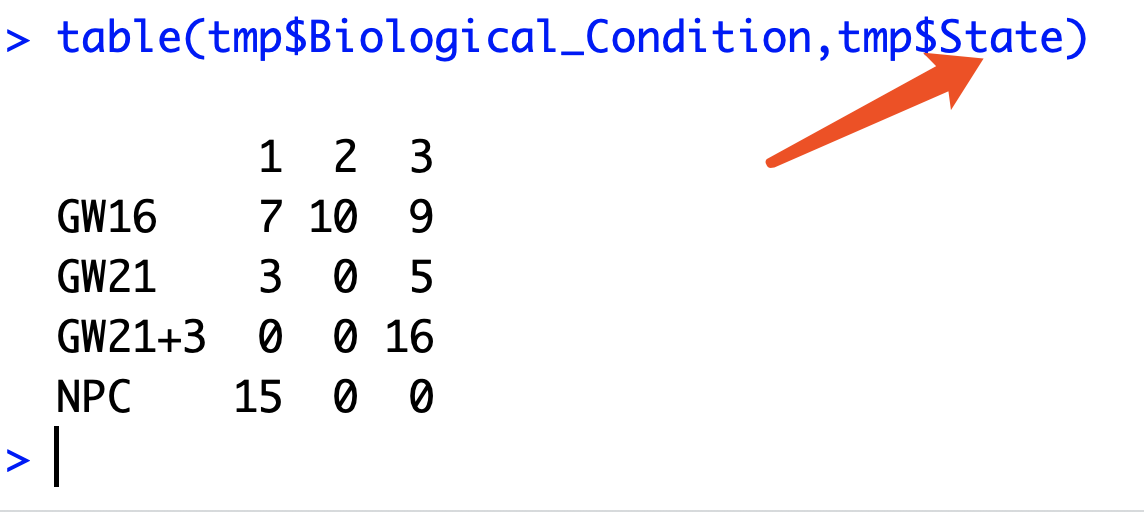
计算Pseudotime得分的10等分里面的不同细胞类型的比例
可以看到这里的State信息并没有用,在学员的求助的文献里面,是把Pseudotime得分进行10等分,因为我们这里只有65个细胞,所以10等分后再看4种细胞的比例,基本上画出来的图会很难看。
不过大体上的思路就是这样咯,十等分是很简单的, 假设得到了,就可以出下面的表格,就是学员想要的条形堆砌图的原始数据了哈。
这个时候,我再留一个疑问,是不是一定要使用monocle来进行细胞谱系推断呢?
文末友情宣传
强烈建议你推荐给身边的博士后以及年轻生物学PI,多一点数据认知,让他们的科研上一个台阶:
- 全国巡讲全球听(买一得五) ,你的生物信息学入门课
- 生信技能树的2019年终总结 ,你的生物信息学成长宝藏
- 2020学习主旋律,B站74小时免费教学视频为你领路