我们在单细胞天地发布的全网第一个单细胞转录组课程,精炼了常规单细胞转录组数据分析主线,就是5大R包, scater,monocle,Seurat,scran,M3Drop,然后10个步骤:
-
step1: 创建对象
-
step2: 质量控制
-
step3: 表达量的标准化和归一化
-
step4: 去除干扰因素(多个样本整合)
-
step5: 判断重要的基因
-
step6: 多种降维算法
-
step7: 可视化降维结果
-
step8: 多种聚类算法
-
step9: 聚类后找每个细胞亚群的标志基因进行亚群命名
-
step10: 继续分类
因为是第一个课程,所以并没有提到单细胞转录组的部分新颖分析要点,比如构建细胞谱系发育,虽然我其实在课程里面也稍微提到过一点,不过怕大家印象不深刻,所以还是有必要单独拿出来讲解一下。
而构建细胞谱系发育,就不得不提Monocle了,值得注意的是有两个版本,我们选择讲解V2,它的官网在:
- 版本2:https://cole-trapnell-lab.github.io/monocle-release/docs/#installing-monocle
- 版本3:https://cole-trapnell-lab.github.io/monocle3/monocle3_docs/
构建细胞谱系发育,也就是pseudotime(拟时序分析),主要是判断不同细胞表达量之间的关系,不同亚群之间表达量过渡的变化就是一条轨迹,类比于随着时间的发育过程的基因表达变化,但并不是真正的时间上的变化,因此我们叫它”拟时序“,而不是”真时间分析“。所以多个细胞亚群可以有多个起点,多条轨迹哦。
同样我们首先拿scRNAseq包的表达矩阵测试
这个包内置的是 Pollen et al. 2014 数据集,人类单细胞细胞,分成4类,分别是 pluripotent stem cells 分化而成的 neural progenitor cells (“NPC”) ,还有 “GW16” and “GW21” ,“GW21+3” 这种孕期细胞,理解这些需要一定的生物学背景知识,如果不感兴趣,可以略过。这个R包大小是50.6 MB,下载需要一点点时间,先安装加载它们。
这个数据集很出名,截止2019年1月已经有近400的引用了,后面的人开发R包算法都会在其上面做测试,比如 SinQC 这篇文章就提到:We applied SinQC to a highly heterogeneous scRNA-seq dataset containing 301 cells (mixture of 11 different cell types) (Pollen et al., 2014).
不过本例子只使用了数据集的4种细胞类型而已,因为 scRNAseq 这个R包就提供了这些,完整的数据是 23730 features, 301 samples, 地址为https://hemberg-lab.github.io/scRNA.seq.datasets/human/tissues/ , 这个网站非常值得推荐,简直是一个宝藏。
这里面的表达矩阵是由 RSEM (Li and Dewey 2011) 软件根据 hg38 RefSeq transcriptome 得到的,总是130个文库,每个细胞测了两次,测序深度不一样。
首先拿到表达矩阵和表型信息
这里仅仅是挑选65个高深度的单细胞文库,代码如下:
library(monocle)
library(scRNAseq)
## ----- Load Example Data -----
data(fluidigm)
ct <- floor(assays(fluidigm)$rsem_counts)
ct[1:4,1:4]
sample_ann <- as.data.frame(colData(fluidigm))
table(sample_ann$Coverage_Type)
table(sample_ann$Biological_Condition)
kp=sample_ann$Coverage_Type=='High'
ct=ct[,kp]
sample_ann=sample_ann[kp,]
然后构建monocle需要的对象
构建Monocle后续分析的所用对象,主要是根据包的说明书,仔细探索其需要的构建对象的必备元素,需要的phenotype data 和 feature data 以及表达矩阵,
注意点: 因为表达矩阵是counts值,所以注意 expressionFamily 参数
gene_ann <- data.frame(
gene_short_name = row.names(ct),
row.names = row.names(ct)
)
pd <- new("AnnotatedDataFrame",
data=sample_ann)
fd <- new("AnnotatedDataFrame",
data=gene_ann)
sc_cds <- newCellDataSet(
ct,
phenoData = pd,
featureData =fd,
expressionFamily = negbinomial.size(),
lowerDetectionLimit=1)
sc_cds
下面是monocle对新构建的CellDataSet 对象的标准操作, 注意estimateDispersions这步的时间和电脑配置密切相关,甚至如果电脑内存不够,还会报错!
sc_cds <- estimateSizeFactors(sc_cds)
sc_cds <- estimateDispersions(sc_cds)
轨迹分析需要指定基因
这些基因可以是细胞亚群直接的差异基因集。
if(F){
disp_table <- dispersionTable(cds)
unsup_clustering_genes <- subset(disp_table,
mean_expression >= 0.1)
cds <- setOrderingFilter(cds, unsup_clustering_genes$gene_id)
dim(cds)
diff_test_res <- differentialGeneTest(cds,
fullModelFormulaStr = "~Biological_Condition")
# 哪怕仅仅是65个单细胞,monocle的这个differentialGeneTest函数运行也不快。
ordering_genes <- row.names (subset(diff_test_res, qval < 0.01))
save(ordering_genes,file = 'ordering_genes_by_Biological_Condition_high.Rdata')
}
load(file = 'ordering_genes_by_Biological_Condition_high.Rdata')
使用官网代码构建谱系发育
cds <- setOrderingFilter(cds, ordering_genes)
plot_ordering_genes(cds)
# 然后降维
cds <- reduceDimension(cds, max_components = 2,
method = 'DDRTree')
# 降维是为了更好的展示数据。
# 降维有很多种方法, 不同方法的最后展示的图都不太一样, 其中“DDRTree”是Monocle2使用的默认方法
# 接着对细胞进行排序
cds <- orderCells(cds)
## 最后两个可视化函数
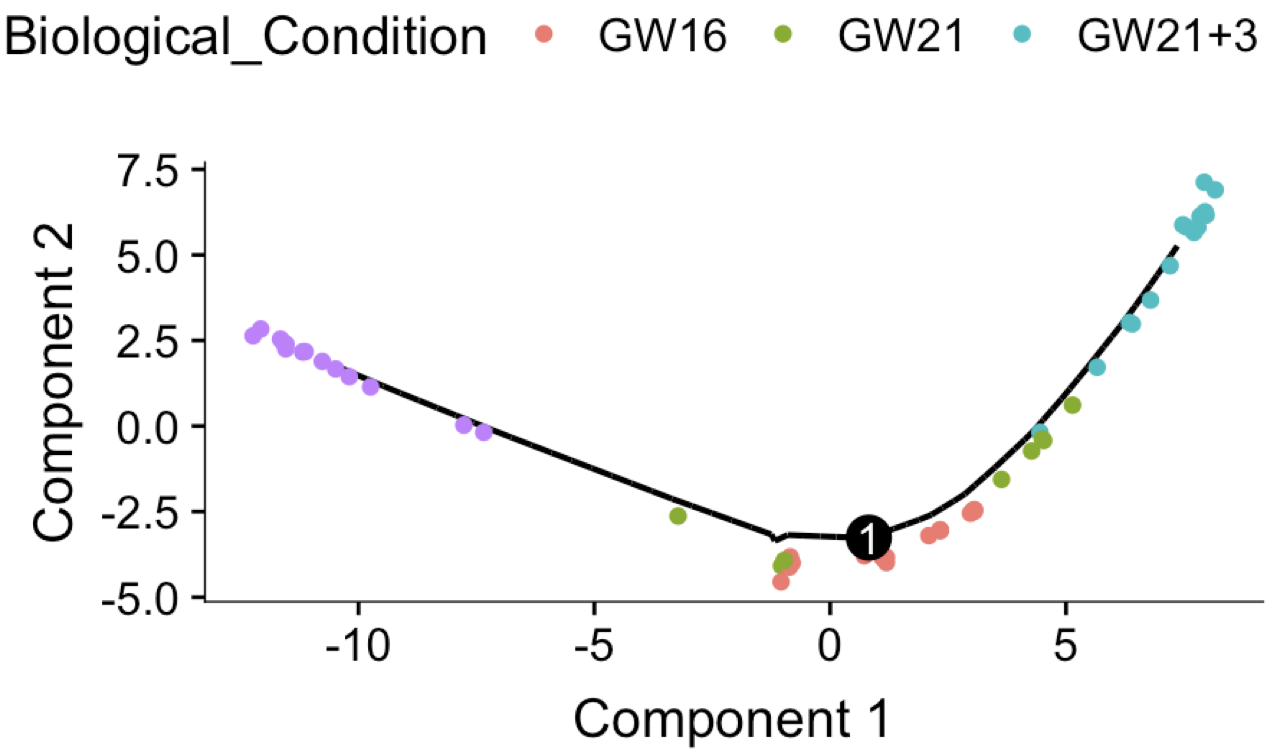
plot_cell_trajectory(cds, color_by = "Biological_Condition")
# 可以很明显看到细胞的发育轨迹
# 还有几个其它可视化函数,我们明天介绍
plot_cell_trajectory(cds, color_by = "State")
plot_cell_trajectory(cds, color_by = "Pseudotime")
plot_cell_trajectory(cds, color_by = "State") +
facet_wrap(~State, nrow = 1)
因为NPC细胞跟另外3种细胞从生理上就不一样,所以是单独的发育轨迹,而 “GW16” and “GW21” ,“GW21+3” 这种孕期细胞,就可以很清晰的看到时间被反映在我们的拟时序分析结果了。
选择错误的基因集去做轨迹分析会怎么样呢?
比如我因为嫌弃monocle的这个differentialGeneTest函数运行太慢,为了简单写教程,就直接直接挑选top2000的MAD基因。
# 加入为了方便起见,直接挑选top2000的MAD基因。
ordering_genes=names(tail(sort(apply(cds@assayData$exprs,1,mad)),2000))
使用这2000个基因去跑拟时序分析,得到的单细胞发育谱系如下:
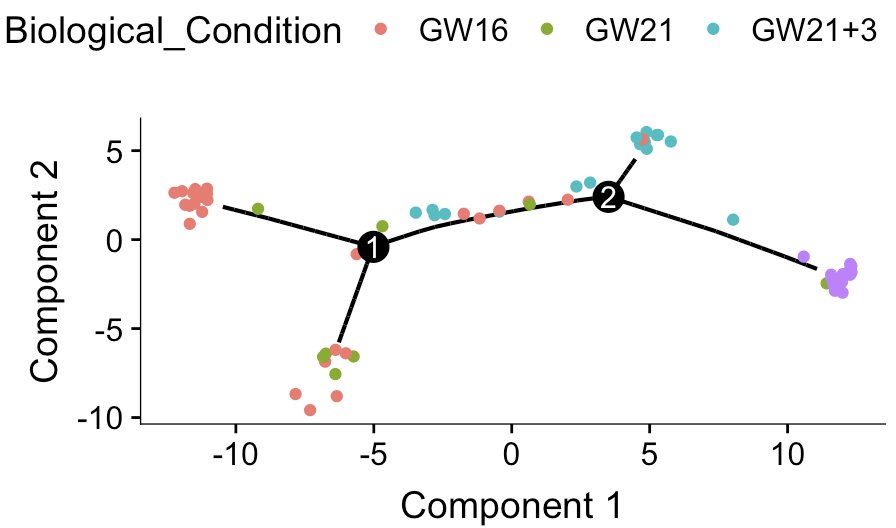
是不是很有趣!
当然了,出这样的图还只是万里长征第一步啦!
下一讲:有了发育轨迹之后呢?
敬请期待
文末友情宣传
强烈建议你推荐给身边的博士后以及年轻生物学PI,多一点数据认知,让他们的科研上一个台阶:
- 全国巡讲全球听(买一得五) ,你的生物信息学入门课
- 生信技能树的2019年终总结 ,你的生物信息学成长宝藏
- 2020学习主旋律,B站74小时免费教学视频为你领路