每月一生信流程栏目灵感来自于《铁汉1991》博客的《每日一生信》,他那个时候介绍的主要是生信基础知识,包括数据结构,数据格式,数据库资源,计算机基础等等,所以每天都可以进步,每天都有成果。这些基础知识已经被分享的七七八八了,所以我这里推陈出新,来一个每月一生信流程,陪生信技能树的粉丝们一起进步!
前面我们推荐两个值得用一个月时间来学习的流程,包括
但是RNA-seq的分析肯定远不止那些啦,拿到基于基因的表达矩阵固然可以根据转录组经典表达量矩阵下游分析大全 里面的R包和代码进行统计可视化,但是表达矩阵并不是凭空产生,上游分析也需要我们有一定的认知,本次我们介绍的流程就会涵盖这些知识点。(很多朋友会下意识的认为RNA-seq数据的上游分析必然是基于Linux,其实也是可以使用bioconductor的全部R包来完成的哦!)
今天学习rnaseqGene
流程的原标题是:RNA-seq workflow: gene-level exploratory analysis and differential expression
- http://www.bioconductor.org/packages/release/workflows/vignettes/rnaseqGene/inst/doc/rnaseqGene.html
这个同样的也没有中文版,讲解了把测序的FASTQ文件比对到参考基因组,或者使用salmon进行定量。介绍了2种得到表达矩阵的方法,涵盖的R包或者软件如下:

在R里面安装这个bioconductor流程
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("rnaseqGene")
因为原文写的实在是太详细了,我这里就不拷贝粘贴了,大家直接去阅读即可
全部目录如下;
- 1 Introduction
- 2 Preparing quantification input to DESeq2
- 3 The DESeqDataSet object, sample information and the design formula
- 4 Exploratory analysis and visualization
- 5 Differential expression analysis
- 6 Plotting results
- 7 Annotating and exporting results
- 8 Removing hidden batch effects(去除批次效应哦!)
- 9 Time course experiments (时间序列转录组数据)
流程代码
我这里其实更加推崇我自己GitHub的airway数据集的分析代码:https://github.com/jmzeng1314/GEO/tree/master/airway_RNAseq 节选如下:
library(airway)
## 表达矩阵来自于R包: airway
# 如果当前工作目录不存在文件:'airway_exprSet.Rdata'
# 就运行下面 if 代码块内容,载入R包airway及其数据集airway后拿到表达矩阵和表型信息
if(!file.exists('airway_exprSet.Rdata')){
library(airway)
data(airway)
exprSet=assay(airway)
group_list=colData(airway)[,3]
save(exprSet,group_list,file = 'airway_exprSet.Rdata')
}
# 大家务必注意这样的代码技巧,多次存储Rdata文件。
# 存储后的Rdata文件,很容易就加载进入R语言工作环境。
load(file = 'airway_exprSet.Rdata')
table(group_list)
# 下面代码是为了检查
if(T){
colnames(exprSet)
pheatmap::pheatmap(cor(exprSet))
group_list
tmp=data.frame(g=group_list)
rownames(tmp)=colnames(exprSet)
# 组内的样本的相似性理论上应该是要高于组间的
# 但是如果使用全部的基因的表达矩阵来计算样本之间的相关性
# 是不能看到组内样本很好的聚集在一起。
pheatmap::pheatmap(cor(exprSet),annotation_col = tmp)
dim(exprSet)
# 所以我这里初步过滤低表达量基因。
exprSet=exprSet[apply(exprSet,1, function(x) sum(x>1) > 5),]
dim(exprSet)
exprSet=log(edgeR::cpm(exprSet)+1)
dim(exprSet)
# 再挑选top500的MAD值基因
exprSet=exprSet[names(sort(apply(exprSet, 1,mad),decreasing = T)[1:500]),]
dim(exprSet)
M=cor(log2(exprSet+1))
tmp=data.frame(g=group_list)
rownames(tmp)=colnames(M)
pheatmap::pheatmap(M,annotation_col = tmp)
# 现在就可以看到,组内样本很好的聚集在一起
# 组内的样本的相似性是要高于组间
pheatmap::pheatmap(M,annotation_col = tmp,filename = 'cor.png')
library(pheatmap)
pheatmap(scale(cor(log2(exprSet+1))))
}
# 以上代码,就是为了检查R包airway及其数据集airway里面的表达矩阵和表型信息
有一个质控的讨论,值得大家看看哦!
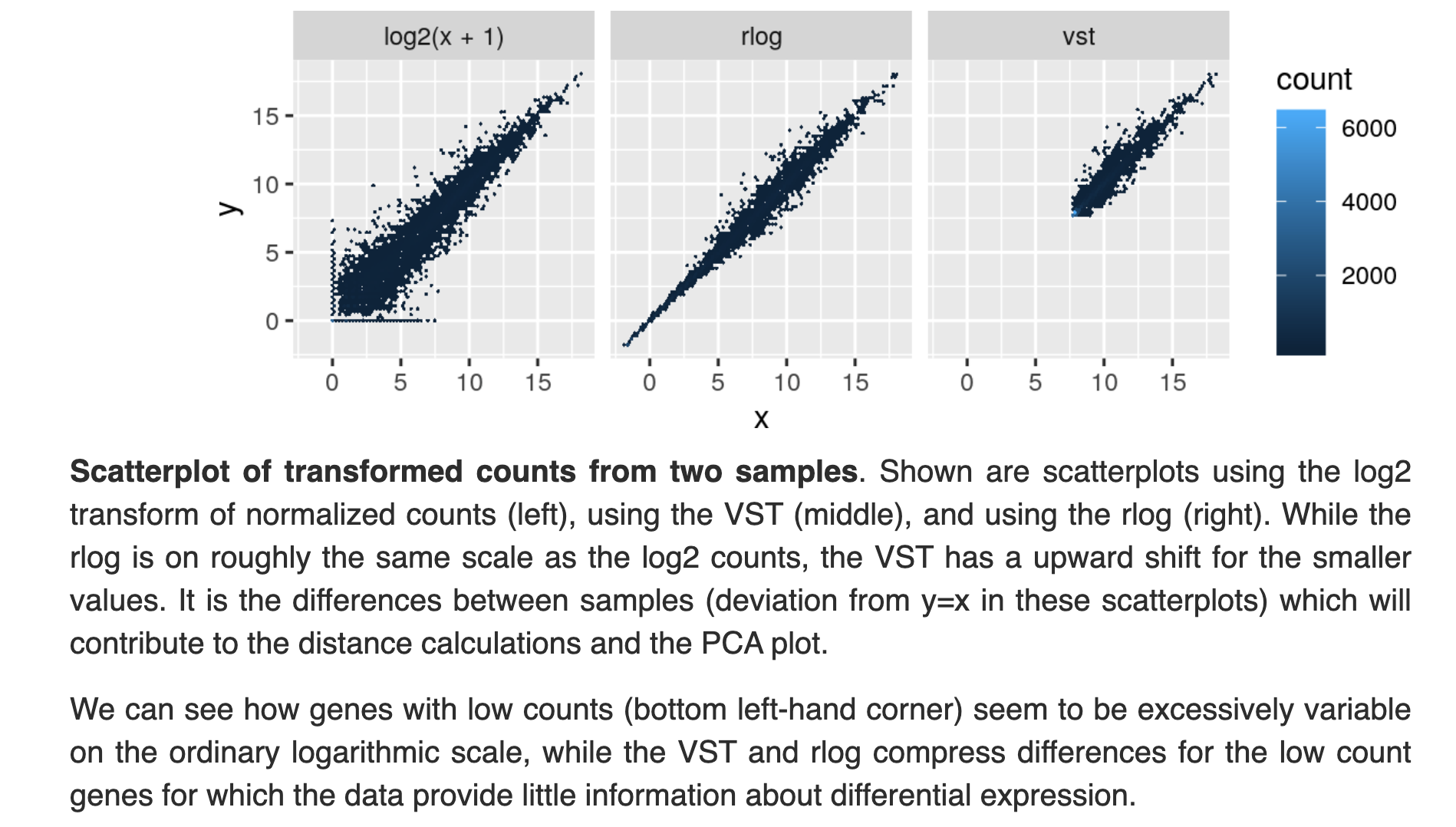
学习这样的流程是需要一定背景知识的
首先是LINUX学习
我在《生信分析人员如何系统入门Linux(2019更新版)》把Linux的学习过程分成6个阶段 ,提到过每个阶段都需要至少一天以上的学习:
- 第1阶段:把linux系统玩得跟Windows或者MacOS那样的桌面操作系统一样顺畅,主要目的就是去可视化,熟悉黑白命令行界面,可以仅仅以键盘交互模式完成常规文件夹及文件管理工作。
- 第2阶段:做到文本文件的表格化处理,类似于以键盘交互模式完成Excel表格的排序、计数、筛选、去冗余,查找,切割,替换,合并,补齐,熟练掌握awk,sed,grep这文本处理的三驾马车。
- 第3阶段:元字符,通配符及shell中的各种扩展,从此linux操作不再神秘!
- 第4阶段:高级目录管理:软硬链接,绝对路径和相对路径,环境变量
- 第5阶段:任务提交及批处理,脚本编写解放你的双手
- 第6阶段:软件安装及conda管理,让linux系统实用性放飞自我
然后是R学习
我在在生信分析人员如何系统入门R(2019更新版) 里面给初学者的知识点路线图如下:
- 了解常量和变量概念
- 加减乘除等运算(计算器)
- 多种数据类型(数值,字符,逻辑,因子)
- 多种数据结构(向量,矩阵,数组,数据框,列表)
- 文件读取和写出
- 简单统计可视化
- 无限量函数学习
必备书籍及视频
书籍贪多不烂,下面2本必买,读5遍以上
《R语言之书-编程与统计》与《Linux 命令行与 shell 脚本编程大全》
视频必须强推生信技能树近30万学习量的基础合辑:
生信技能树关于RNA-seq上下游数据分析的教程的确不少了
因为做目录确实很浪费时间,差不多就下面这些,大家先学习吧:
- 转录组经典表达量矩阵下游分析大全
- 原创10000+生信教程大神给你的RNA实战视频演练
- 转录组经典表达量矩阵下游分析大全
- 可变剪切,差异转录本,或者差异外显子流程大全
- RNA测序究竟有多可靠呢
- TCGA数据库里面的乳腺癌样本RNA-seq数据是配对的有哪些?
- 生信小白的RNA-seq实战历程
- RNA-seq数据分析指南
后记
听说隔壁openbiox团队在组织翻译这个bioconductor流程系列,而且还是由我们生信技能树元老-思考问题的熊领头,希望他们的翻译成果早日出版!