众所周知,癌症具有异质性,在乳腺癌领域,不同亚型的癌症比不同器官来源癌症的差异要大很多。最简单癌症分类,当然是一个基因,比如ER阳性或者ER阴性的乳腺癌患者,并不是说人类有2万多个蛋白编码基因就可以有2万多种分类,其实在乳腺癌领域常用的分类,就是ER,HER2,PR等等,如果这3个基因都不表达,就是临床里面比较恶性的三阴性乳腺癌啦。
如果你并不研究乳腺癌,你可能会思考,如果是根据3个基因的表达量高低,比如ER,HER2,PR,那么应该是2X2X2=8种分类,但是实际上通过IHC分型(就是检查具体基因表达量)方法,乳腺癌被划分为激素受体(ER、PR)阳性组和阴性组,然后各自划分2个组,也就是4个组:
- 激素受体(ER、PR)阳性组
- 管腔A型(ER阳性,PR≥20%,HER2阴性,Ki-67<20%)
- 管腔B型(ER阳性,PR<20%和/或HER2阳性和/或Ki-67≥20%)
- 激素受体(ER、PR)阴性组
- HER2过表达型(ER阴性,PR阴性,HER2阳性)
- 基底样型(ER阴性,PR阴性,HER2阴性)
可以看到,这些具体的检测指标是具有一定的可操作空间,灵活性,而且IHC检测的是蛋白表达水平,而我们通常是拿到的mRNA层次的表达矩阵,阳性和阴性这样的二元分类概念会量化成为了具体的表达量数值。而根据表达矩阵来进行癌症亚型的划分,就是分子分型(GEP),比如基于PAM50的GEP分型可将乳腺癌分为不同的亚型,包括表达雌激素受体(ER)相关因子的亚型(管腔型)、表达人类表皮生长因子受体2(HER2)相关通路因子的亚型(HER2过表达亚型)和表达基底因子但不表达激素受体通路的基底样乳腺癌(BLBC)亚型。
考虑到乳腺癌领域应用最广泛的就是基于PAM50的GEP分型,有必要系统性学习一下它的分型原理。
50个基因的来龙去脉
Parker et al (2009) published microarray data (GEO data set GSE10886),数据可以比较方便的下载,重现整个分析思路也不难,简单理解一下:
- 4个基因表达芯片数据集,定义了“intrinsic” gene set ,有1906个基因
- 对189病人的1906个基因的表达矩阵层次聚类(median centered by feature/gene, Pearson correlation, average linkage)
- 使用SigClust对层次聚类结果划分cluster
- SigClust得到9个cluster,包含 luminal A (dark blue), luminal B (light blue), HER2-enriched (pink), normal-like (green), and basal-like (red).
- 然后精简基因集,每个分组选择top10的基因,得到5个分组的共50个基因。这里使用了Classification by Nearest Centroids (ClaNC) 算法。
- 也就是说189个病人里面的122个划分结果与qRT-PCR 一致。
如下所示:

因为有了PAM50,所以以后临床实践只需要测定50个基因的表达量即可对病人进行初步分类。而且作者提供了全套代码和数据:
-
The centroids, gene lists, and R code to produce the classification are all available along with the clinical information for the training set on this page: https://genome.unc.edu/pubsup/breastGEO/
-
Specifically, the R code and supporting data files are here: https://genome.unc.edu/pubsup/breastGEO/PAM50.zip
-
And the centroids alone are here: https://genome.unc.edu/pubsup/breastGEO/pam50_centroids.txt
那么如何使用这50个基因的分类器呢?
PAM50分子分型
step1:拿到50个基因在所有样本的表达量矩阵
比如作者给出的例子是:”220arrays_nonUBCcommon+12normal_50g.txt”文件,很明显,有233个样本啦!
trainFile='220arrays_nonUBCcommon+12normal_50g.txt'
phe=t(read.table(trainFile,sep = '\t',row.names = 1)[1:2,])
head(phe)
x<-read.table(trainFile,skip = 2,sep = '\t',row.names = 1)
x[1:4,1:4]
colnames(x)=phe[,1]
dim(x)
# 因为是 "220arrays_nonUBCcommon+12normal_50g.txt", 所以是232个样本:
boxplot(x[,1:4])
standardize<-function(x){
annAll<-dimnames(x)
x<-scale(x)
dimnames(x)<-annAll
return(x)
}
x<-standardize(x)
x[1:4,1:4]
boxplot(x[,1:4])
# 表达矩阵被zscore后,数据量集中在0附近
step2:读取分类器
trainCentroids='pam50_centroids.txt'
# load the published centroids for classifcation
pamout.centroids<-read.table(trainCentroids,sep="\t",header=T,row.names=1)
head(pamout.centroids)
# pam50的5个亚型的50个基因的
boxplot(pamout.centroids)
pheatmap::pheatmap(pamout.centroids)
rowSums(pamout.centroids)
step3: 把表达矩阵和分类器进行比较
这个时候有3种比较的算法,就是euclidean距离,以及pearson或者spearman相关系数!载入前面处理好的分离器和表达矩阵,然后简单比较即可:
rm(list = ls())
options(stringsAsFactors = F)
library(ctc)
load(file = 'input_for_pam50.Rdata')
overlapSets<-function(x,y){
# subset the two lists to have a commonly ordered gene list
x<-x[dimnames(x)[[1]] %in% dimnames(y)[[1]],]
y<-y[dimnames(y)[[1]] %in% dimnames(x)[[1]],]
#and sort such that thing are in the correct order
x<-x[sort.list(row.names(x)),]
y<-y[sort.list(row.names(y)),]
return(list(x=x,y=y))
}
temp <- overlapSets(pamout.centroids,x)
# 如果表达矩阵和分类器共有基因少于50个,要注意一下。
centroids <- temp$x
tmp <- temp$y
library("impute")
dat=t(as.data.frame(impute.knn(as.matrix(t(temp$y)))$data))
# 200个样本的表达矩阵,去PAM50矩阵进行对比计算
d1=apply(centroids,2,function(i){
# dist 函数返回倒三角矩阵
dist(t(cbind(i,dat)))[1:ncol(dat)]
})
d2=cor(dat,centroids,method="pearson",use="pairwise.complete.obs")
d3=cor(dat,centroids,method="spearman",use="pairwise.complete.obs")
p1=apply(d1,1, function(x){
colnames(d1)[which.min(x)]
})
p2=apply(d2,1, function(x){
colnames(d2)[which.max(x)]
})
p3=apply(d3,1, function(x){
colnames(d3)[which.max(x)]
})
table(p1,p2)
table(p3,p1)
table(p3,p2)
save(prediction,distances,centroids,dat,file = 'output_for_pam50.Rdata')
step4:检查分离器效果
rm(list = ls())
options(stringsAsFactors = F)
library(ctc)
load(file = 'input_for_pam50.Rdata')
load(file = 'output_for_pam50.Rdata')
table(p1)
phe=as.data.frame(cbind(phe,p1,p2,p3))
colnames(phe)=c('sample','type','dist','pearson','spearman')
phe=phe[phe$type!='',]
table(phe$type,phe$dist)
table(phe$type,phe$pearson)
table(phe$type,phe$spearman)
step5: 把PAM50应用到你自己的数据
rm(list = ls())
options(stringsAsFactors = F)
library(ctc)
load(file = 'input_for_pam50.Rdata')
if(F){
# 这里面的数据集文件有点大, 所以我注释掉这一块代码
# [AgilentG4502A_07_3]
# (n=597) TCGA hub
# 下载 AgilentG4502A_07_3 后自己格式化成为breat_array_tcga.Rdata
load('breat_array_tcga.Rdata')
cg=rownames(pamout.centroids)
cg
cg=cg[cg %in% rownames(breat_array_tcga)]
cg
dat=breat_array_tcga[cg,]
centroids=pamout.centroids[cg,]
save(centroids,dat,file = 'input_for_pam50_tcga_array.Rdata')
}
load(file = 'input_for_pam50_tcga_array.Rdata')
d1=apply(centroids,2,function(i){
# dist 函数返回倒三角矩阵
dist(t(cbind(i,dat)))[1:ncol(dat)]
})
p1=apply(d1,1, function(x){
colnames(d1)[which.min(x)]
})
table(p1)
pid=gsub('[.]','-',substring(colnames(dat),1,12))
nt=substring(colnames(dat),14,15)
aphe=data.frame(pid,nt,p1)
aphe=aphe[aphe$nt=='01',]
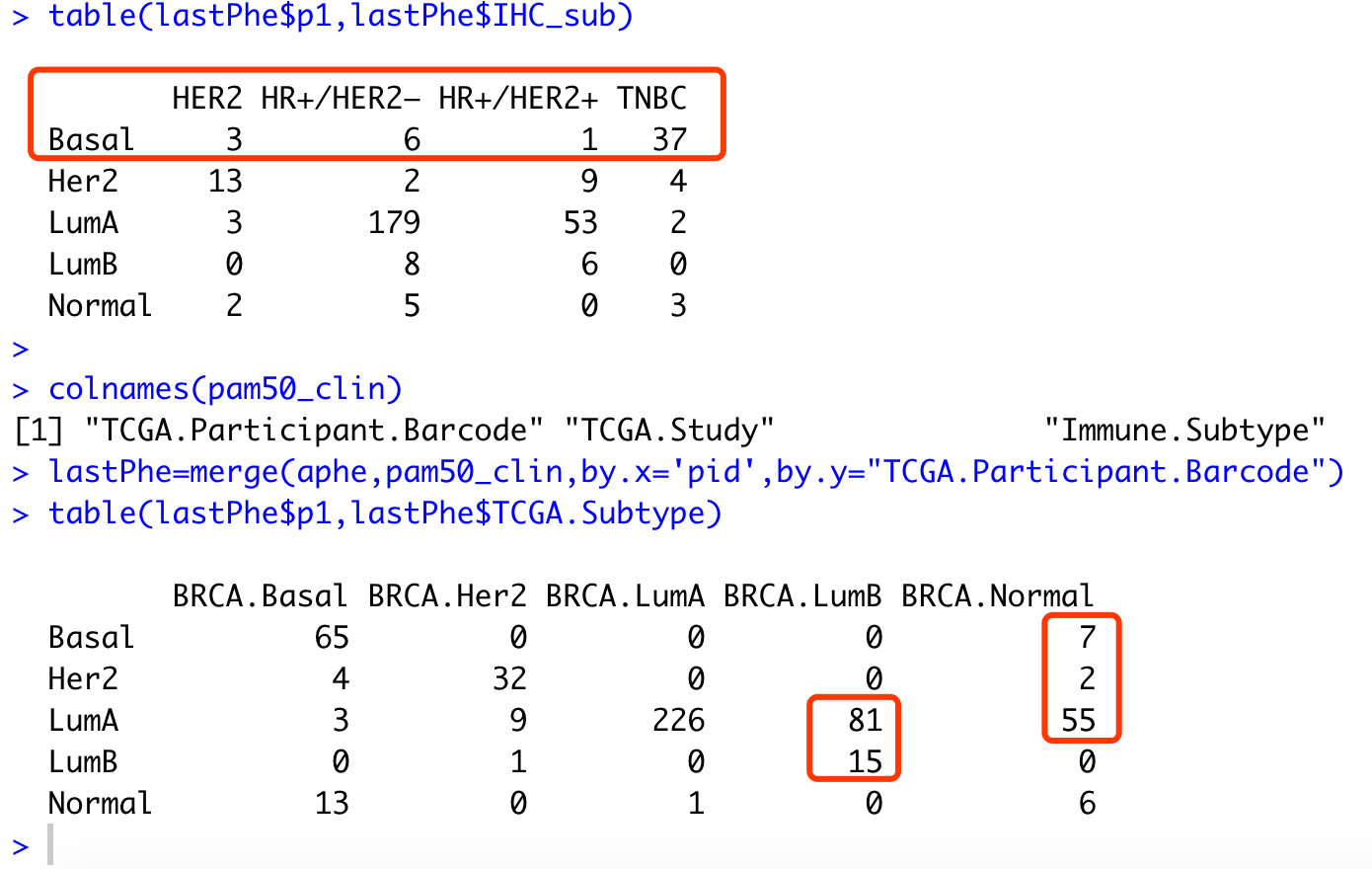
load('subtype_anno.Rdata')
lastPhe=merge(aphe,IHC_clin,by.x='pid',by.y='id')
table(lastPhe$p1,lastPhe$IHC_sub)
colnames(pam50_clin)
lastPhe=merge(aphe,pam50_clin,by.x='pid',by.y="TCGA.Participant.Barcode")
table(lastPhe$p1,lastPhe$TCGA.Subtype)
可以看到TNBC和basel的重合度非常高,然后LumA和Normal其实是很难区分开来的。
全部代码和数据,都整理好啦!需要付费获取,供有需要的朋友!
文末友情宣传
强烈建议你推荐给身边的博士后以及年轻生物学PI,多一点数据认知,让他们的科研上一个台阶:
- 底裤价转录组产品线(还送数据分析培训)(八九百一个样品)
- 三维基因组学分析实战培训班,线上直播课,2天仅需399(生信技能树粉丝特权价格)
- 生信技能树的2019年终总结 ,你的生物信息学成长宝藏
- 2020学习主旋律,B站74小时免费教学视频为你领路
付费内容分割线
为有效杜绝黑粉跟我扯皮,设置一个付费分割线,这样它们就没办法复制粘贴我的代码,也不可能给我留言骂街了!(这个代码含金量很高,需要付费18元,才能拿到测试数据和代码)
另外,大家不要随意付费施舍我,没有必要请不要购买,这个代码对需要它的人来说,至少值几百块钱!如果你确实想感激我,可以去生信技能树次页找到我的原创推文进行赞赏,赞赏是无私的,这个付费代码是商业活动,请不要随便参与!
综合脚本
链接在:链接:https://share.weiyun.com/59b2Xn3
这个微云链接需要密码,付费18元可以得到,请移步到微信公众号: https://mp.weixin.qq.com/s/7C4LnLf7bSP1zFPM7RRS3g 付费获取哈!
PAM50有一些临床应用
其中nanostring公司提出:The Prosigna Breast Cancer Prognostic Gene Signature Assay
链接是:https://www.nanostring.com/diagnostics/prosigna/technology/prosigna-algorithm

PAM50的基因数量可以精简
2011年一篇文章: 提出了Montreal cohort of 87 patients的30个基因,也可以比较好的区分这些亚型,Of these 30 genes, only 7 of the 30 genes overlapped with the PAM50 (FOXA1, MLPH, ESR1, SLC39A6, NAT1, GRB7 and ERBB2) (Parker et al, 2009)