在分享我是如何培养学徒的时候,提到了一个很有意思的数据处理故事:
- 七步走纯R代码通过数据挖掘复现一篇实验文章(第七步WGCNA)
- 你要挖的公共数据集作者上传了错误的表达矩阵肿么办(如何让高手心甘情愿的帮你呢?)
- 你可能不适合做人(学徒给我的6个暴击)
就是这个 你可能不适合做人(学徒给我的6个暴击),学徒在完成salmon流程走一波airway这个转录组数据集拿到表达矩阵,跟我以前表达矩阵,联合起来看相关性。结果发现了两个矩阵差异非常大,不符合认知,因为同样的样本走不一样的流程,理论上表达矩阵并不会发生很大的差异。所以当时我就认为他数据处理有问题,现在看来,要给他道歉。
实际上,我今天跟我们生信技能树讲师团队重新讨论这个相关性问题的时候,我发现了其实不同的流程会带上一个类似于“批次效应”的东西,这个效应甚至大于样本之间的差异,如果你选择全局基因表达量来看的话。我们现在就来一步步看问题所在,需要先明确几个变量- 走转录组定量流程,gtf文件里面有五六万的基因
- 过滤低表达量后,基因数量仍然是2万左右
- 两个不同转录组定量流程影响相关性,称为流程效应
- 两个分组(肿瘤和对照)当然相关性不一样,称为分组效应
- 每个样本都会有不一样的地方,称为个体效应
首先查看原始counts值的相关性
> overM=cbind(ensembl_matrix[overg,],mat[match(overg,d[,2]),]) > overM[1:4,1:4] SRR7707733 SRR7707734 SRR7707735 SRR7707736 MT-RNR1 6647 995 909 2109 MT-RNR2 23850 3310 3667 7389 MT-ND1 345 226 293 199 MT-ND2 522 272 390 234 > pheatmap::pheatmap(cor(overM)) >得到的相关性图如下:
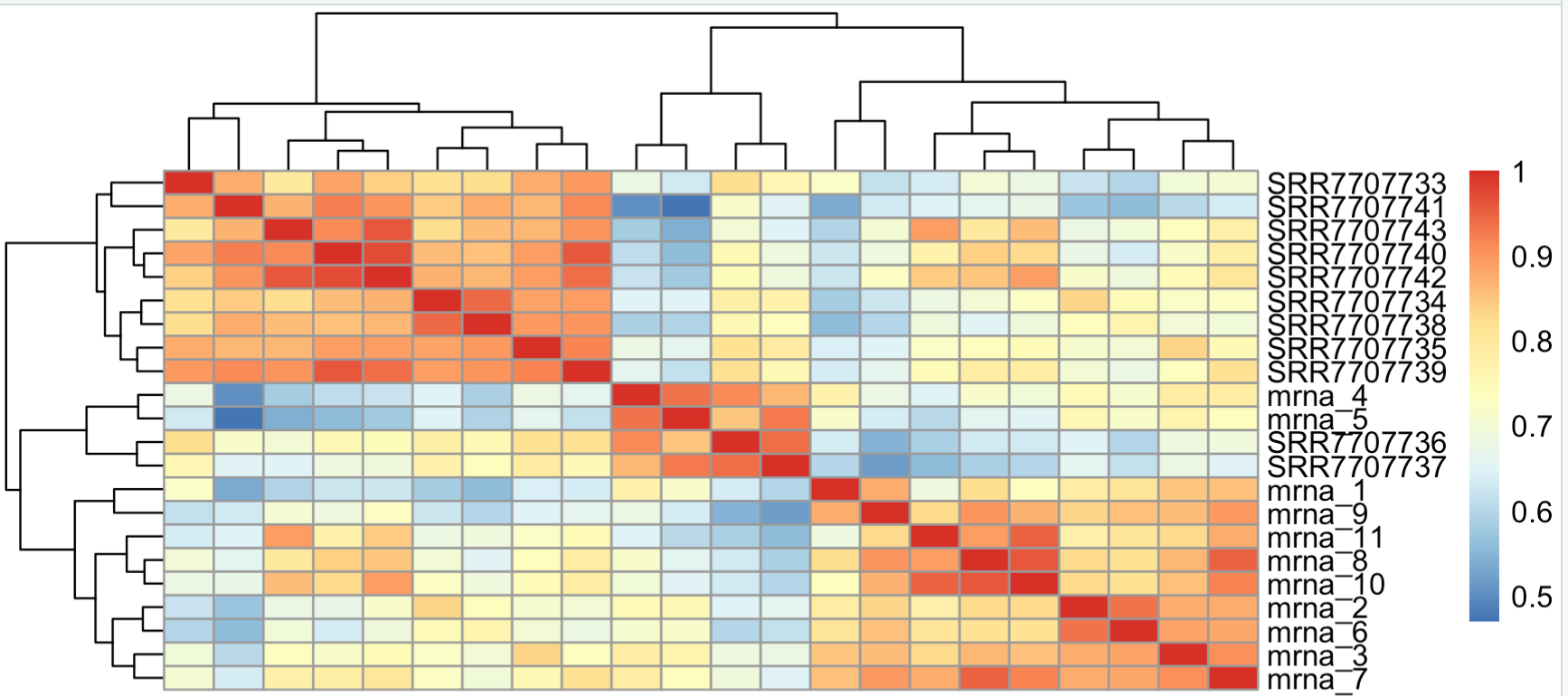
可以看到,流程之间的差异很大,同样的样本在两个不同流程拿到的表达矩阵相关性并没有高于该样本与其它样本的相关性。这个其实是因为纯粹的counts矩阵,并不适合来计算相关性,取一下log转换比较好。log转换后的counts矩阵
代码如下:
pheatmap::pheatmap(cor(log2(overM+1)))可以看到,仅仅是log转换,相关性就提高了很多,出图:
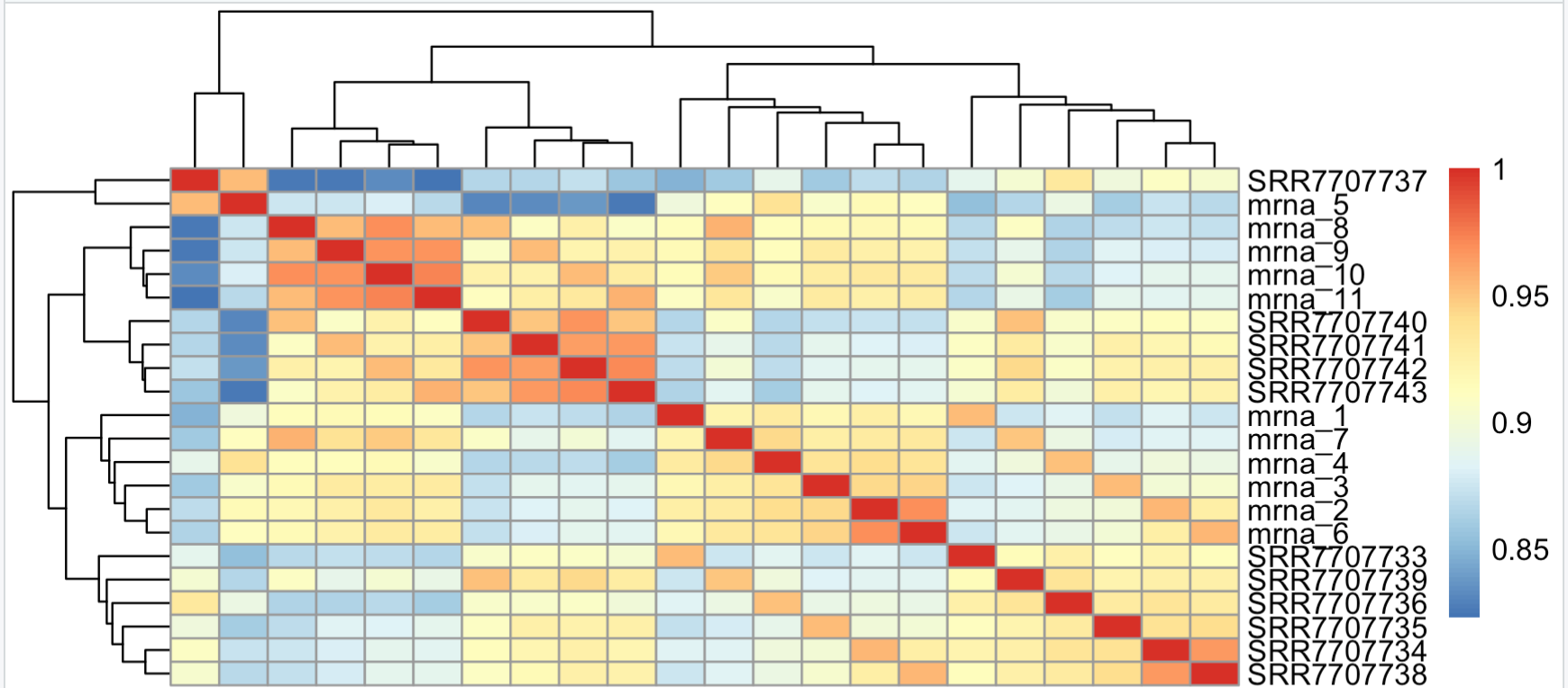
这个时候,虽然流程的差异是存在的,但是分组的差异成为了第一要素。
值得注意的是,并不是每一次分组效应都能显示出来,我们的示例里面是肿瘤和对照,当然是分组效应很明显,有时候大家的项目可能是加药处理前后,万一你的药物没有效果,那么分组效应就不存在的哈。我们取cpm后的矩阵
cpm仅仅是针对文库大小的效应去除掉。
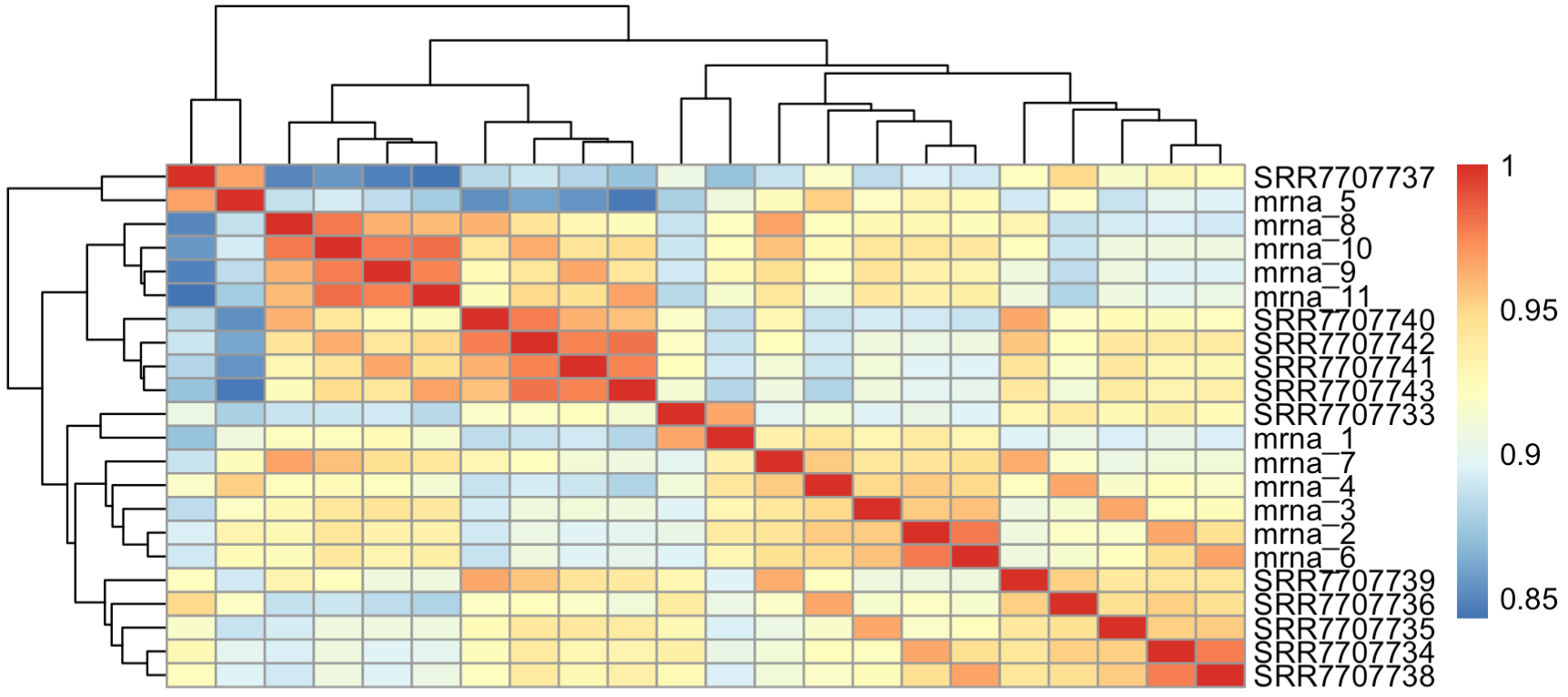
cpmM=log(edgeR::cpm(overM)+1) pheatmap::pheatmap(cor(cpmM))可以看到,跟前面的log的counts矩阵出图类似,说明文库大小并不会显著的影响样本之间的表达矩阵相关性哦。
选取top500的mad基因
首先按照两个表达矩阵合并后的矩阵来挑选top500的mad基因,代码如下:
highM=cpmM[names(tail(sort(apply(cpmM,1,mad)),500)),] pheatmap::pheatmap(cor(highM))有意思的就来了,可以看到,两个流程的效应又一次大于分组效应。因为我们挑选到的基因就是在两个不同流程定量有差异的基因,相当于是放大了流程效应。
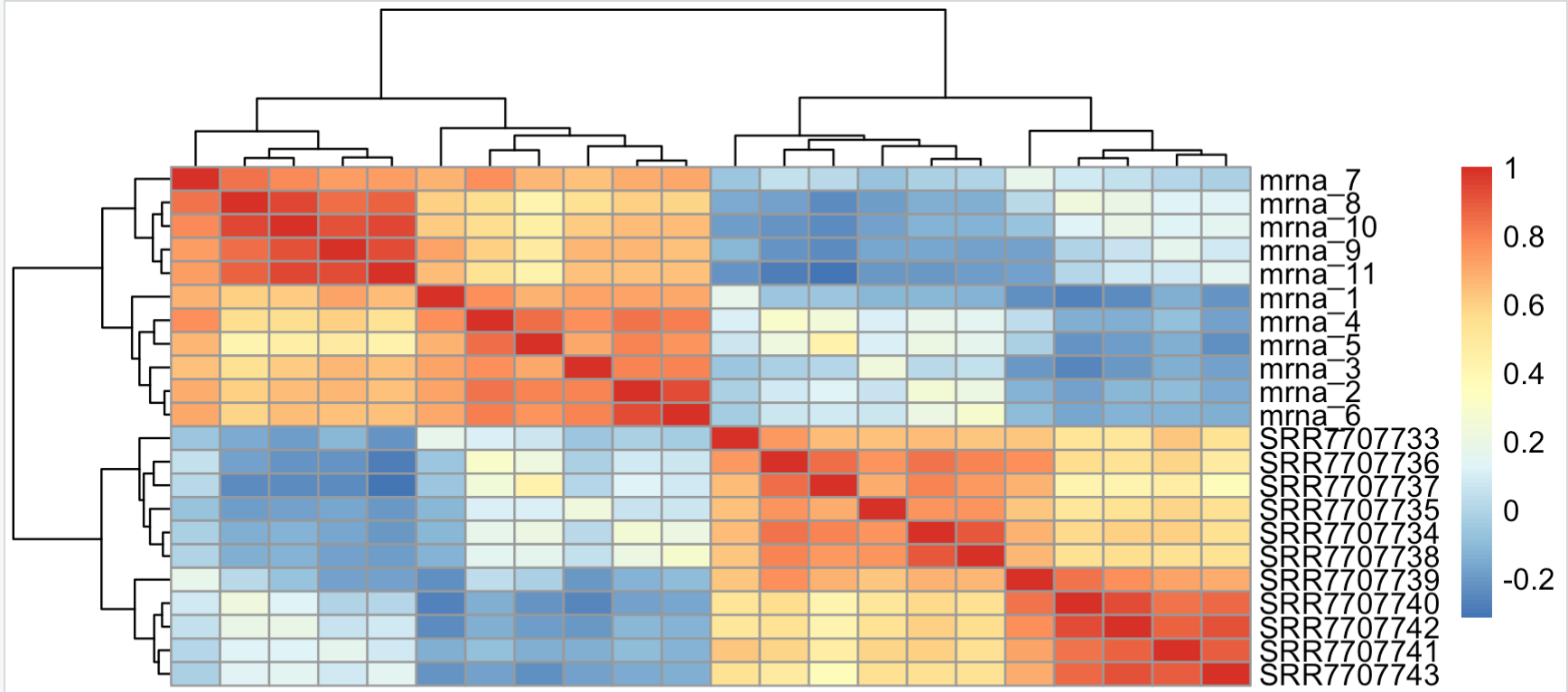
然后我们独立选取各自的top500的mad基因,再去交集hg1=names(tail(sort(apply(cpmM[,1:11],1,mad)),500)) hg2=names(tail(sort(apply(cpmM[,12:22],1,mad)),500)) hg=intersect(hg1,hg2) hgM=cpmM[hg,] pheatmap::pheatmap(cor(hgM))有意思的又来了,两个不同流程各自的top500的mad基因有443个交集,说明那些表达量动态变化的基因在不同流程仍然是趋势一致。
如下所示,流程效应被淹没了,分组效应被体现出来了,个体效应在normal组没有太大的差异,但是在tumor组里面差异蛮大的,说明了肿瘤本身的异质性。
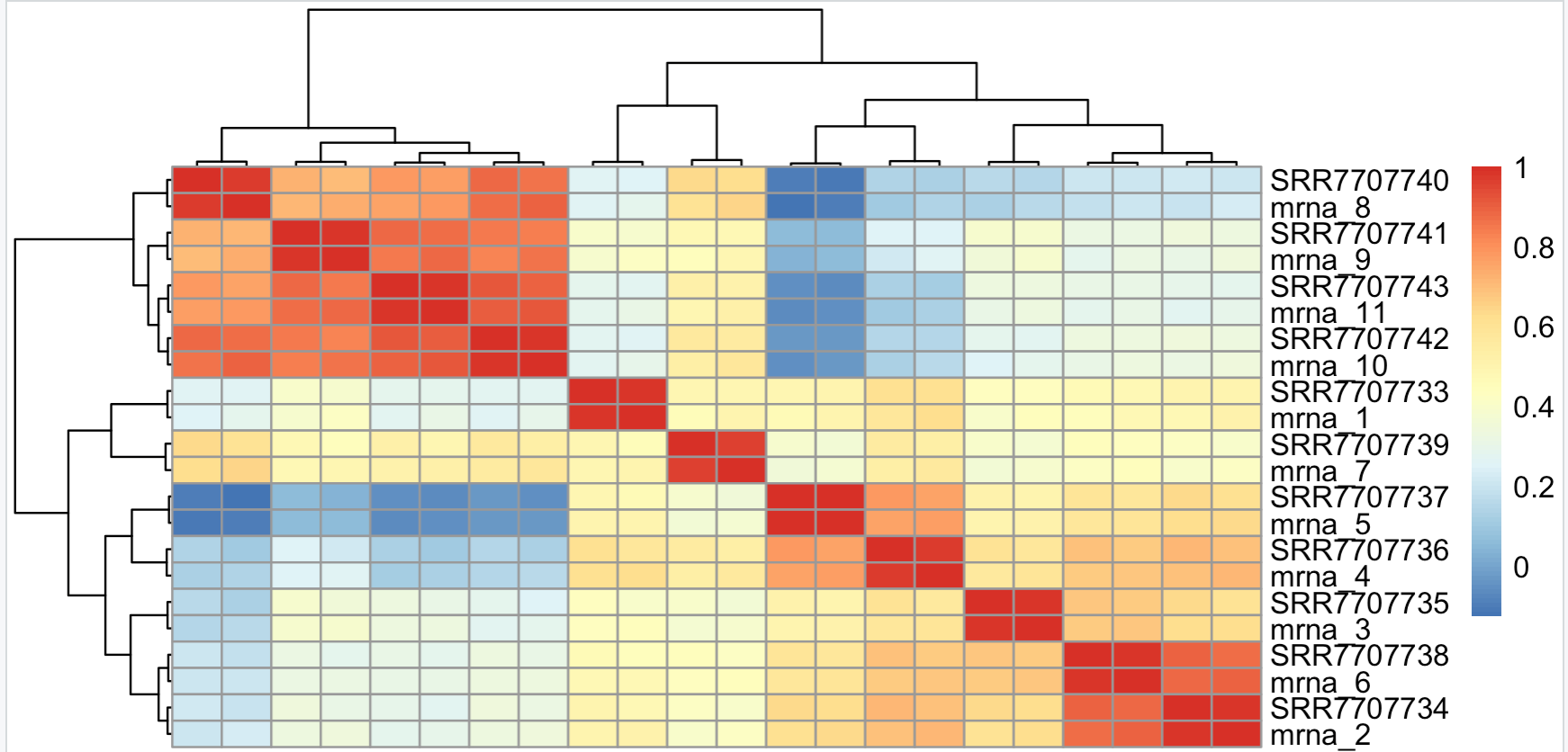
是不是很有意思,对表达矩阵的认识又深入了一些。文末友情宣传
强烈建议你推荐我们生信技能树给身边的博士后以及年轻生物学PI,帮助他们多一点数据认知,让科研更上一个台阶:
- 生信爆款入门-全球听(买一得五)(第4期),你的生物信息学入门课
- 数据挖掘第2期(两天变三周,实力加量),医学生/临床医师首选技能提高课
- 生信技能树的2019年终总结 ,你的生物信息学成长宝藏
- 2020学习主旋律,B站74小时免费教学视频为你领路,还等什么,看啊!!!