对人和鼠科研项目来说,各种NGS测序数据分析起来都是有成熟的流程和工具,但是并不是使用的物种都是有参考基因组和注释文件的,走流程的前提就需要先测定自己研究的物种的基因组,组装起来。
比如最近有粉丝咨询肺吸虫(Paragonimus westermani)也称“卫氏并殖吸虫”的转录组数据分析,我就顺手查了一下,发现ensembl等数据库并没有它的参考基因组信息。其中 2014 Aug 12. doi: 10.7717/peerj.484 发表了它的线粒体基因组,然后 January 2019, giy146, https://doi.org/10.1093/gigascience/giy146 有它的全基因组信息。
恰好这篇文章比较好的展现了一个新物种参考基因组组装的科研项目思路:
- We sequenced and assembled the genome of P. westermani, which is among the largest of the known pathogen genomes with an estimated size of 1.1 Gb.
- A 922.8 Mb genome assembly was generated from Illumina and Pacific Biosciences (PacBio) sequence data, covering 84% of the estimated genome size.
- The genome has a high proportion (45%) of repeat-derived DNA, particularly of the long interspersed element and long terminal repeat subtypes, and the expansion of these elements may explain some of the large size.
- We predicted 12,852 protein coding genes, showing a high level of conservation with related trematode species.
- The majority of proteins (80%) had homologs in the human liver fluke Opisthorchis viverrini, with an average sequence identity of 64.1%. Assembly of the P. westermani mitochondrial genome from long PacBio reads resulted in a single high-quality circularized 20.6 kb contig.
- The contig harbored a 6.9 kb region of non-coding repetitive DNA comprised of three distinct repeat units.
- Our results suggest that the region is highly polymorphic in P. westermani, possibly even within single worm isolates.
就是物种基因组大小预估,测序组装,重复序列,蛋白编码基因分析,物种进化分析。
测序数据如下:

可以看到这个策略里面有pacbio,但是并没有10x和hic哈,属于比较简单的测序。
组装效果评价:
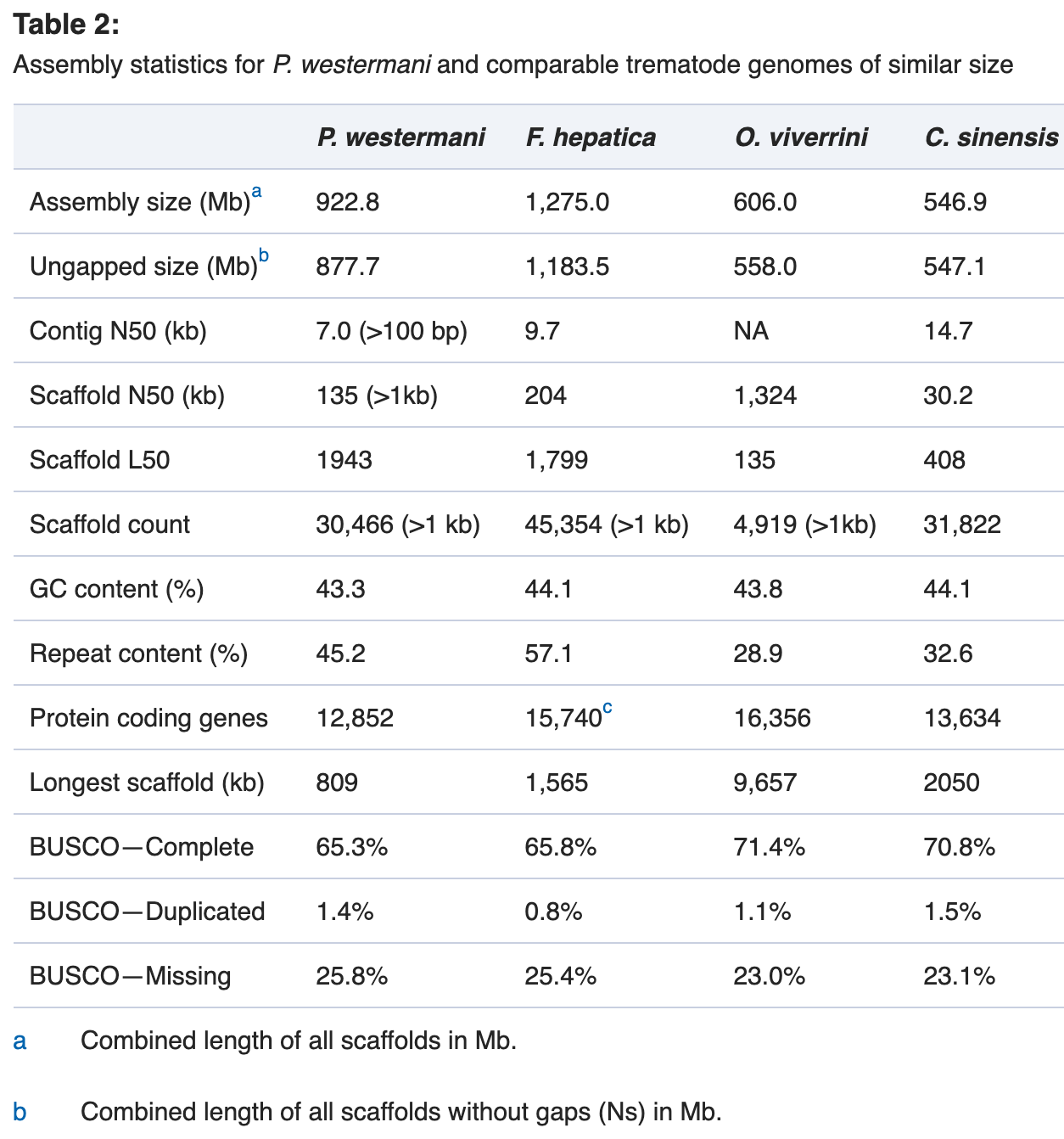
可以看到,组装结果一般般吧。我看最近的雷公藤通过 PacBio,10×Genomics 测序,研究者获得了高质量的雷公藤基因组(~365.95Mb,Contig N50=4.36Mb),结合 Hi-C 数据,将~315.08Mb 的序列挂载到23条染色体上,Scaffold N50=13.52Mb。
文章题目:Genome of Tripterygium wilfordii and identification of cytochrome P450 involved in triptolide biosynthesis
发表期刊:Nature Communication
发表时间:2020.02.20
研究材料:Tripterygium wilfordi
测序策略:PacBio (~207×), 10×Genomics (~327×), Hi-C
真的是差距不是一点点啊!!!没有10x和hic的基因组组装效果简直没法看。在Nature Communication杂志还可以看到很多新物种基因组组装,包括白羽扇豆染色体水平高质量基因组,基因组大小为~558.74Mb,Contig N50 = 1.76Mb,Scaffold N50 = 18.66Mb,注释得到48,719个蛋白编码基因。还有线虫的研究,通过 PacBio 和 Hi-C 测序技术,生成 C. remanei 染色体水平的基因组(~124.8Mb),Contig N50 = 4.042Mb,Scaffold N50 =21.502Mb,利用 BUSCO 评估基因组完整性为~97.9%,注释得到26,308个蛋白编码基因。
重复序列鉴定,蛋白编码基因鉴定,已经物种进化关系分析大家就自行查看文献了哈。
最后给大家推荐一个物种测序策略:
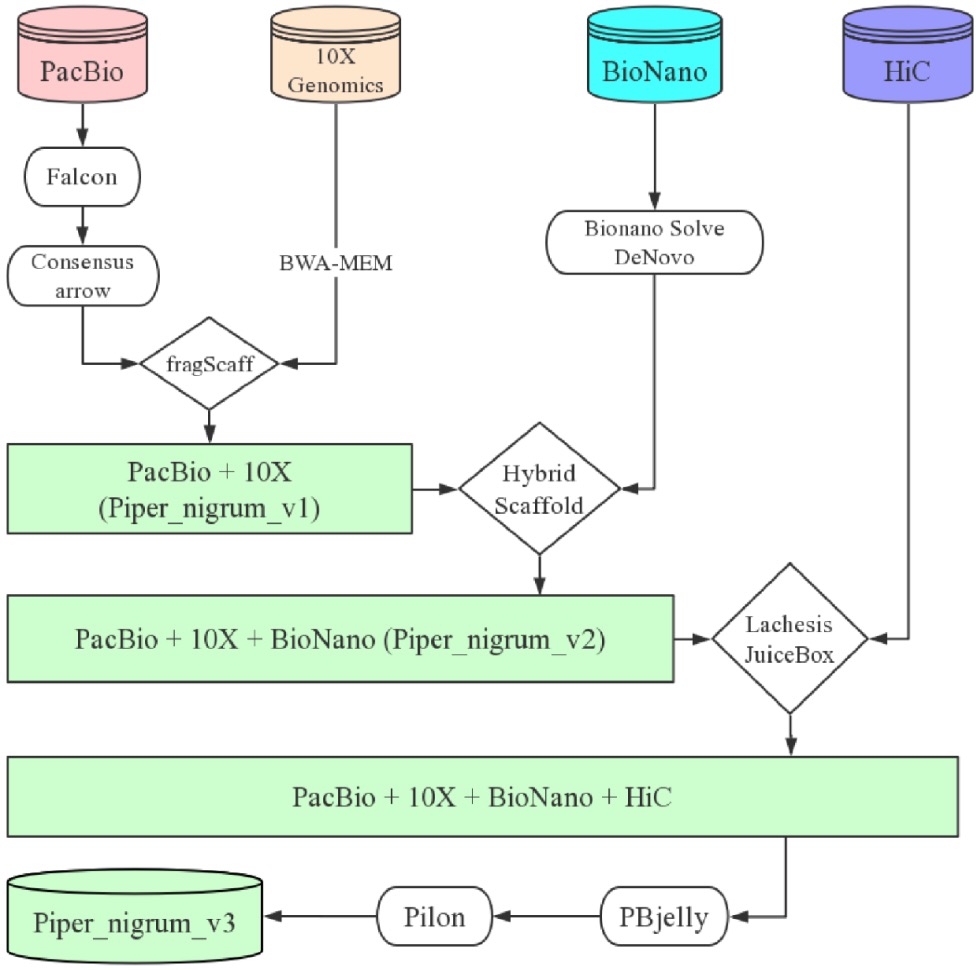
各大科研服务公司都提供这样的服务,费用取决于物种的基因组大小,测序的精细程度,约5-10万左右就可以拿下一个新物种的基因组组装哦!文末友情宣传
强烈建议你推荐我们生信技能树给身边的博士后以及年轻生物学PI,帮助他们多一点数据认知,让科研更上一个台阶:
- 生信爆款入门-全球听(买一得五)(第4期),你的生物信息学入门课
- 数据挖掘第2期(两天变三周,实力加量),医学生/临床医师首选技能提高课
- 生信技能树的2019年终总结 ,你的生物信息学成长宝藏
- 2020学习主旋律,B站74小时免费教学视频为你领路,还等什么,看啊!!!